Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
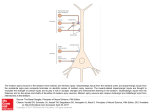
Naⴙ/Ca2ⴙ Exchanger Maintains Ionic Homeostasis in the Peri-Infarct Area Anna Tortiglione, PhD; Barbara Picconi, PhD; Ilaria Barone, PhD; Diego Centonze, MD; Silvia Rossi, MD; Cinzia Costa, MD; Massimiliano Di Filippo, MD; Alessandro Tozzi, PhD; Michela Tantucci, PhD; Giorgio Bernardi, MD; Lucio Annunziato, MD; Paolo Calabresi, MD Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 Background and Purpose—A prominent feature of cerebral ischemia is the excessive intracellular accumulation of both Na⫹ and Ca2⫹ ions, which results in subsequent cell death. The plasma membrane Na⫹/Ca2⫹ exchanger (NCX), regulates the distribution of these ions acting either in the forward mode or in its reverse mode and it can play a critical role in brain ischemia. However, it is unclear whether the activity of NCX leads to detrimental or beneficial effects. Methods—Extracellular field potentials and whole-cell patch clamp recordings were obtained from rat corticostriatal brain-slice preparations in the peri-infarct area 24 hours after the permanent middle cerebral artery occlusion. Ischemia was induced in rats by permanents middle cerebral artery occlusion. Results—Bepridil, an inhibitor of NCX, reduced in a concentration-dependent manner (IC50⫽68 mol/L) the field potential amplitude recorded from the peri-infarct area of corticostriatal slices. Conversely, no change was observed in sham-operated animals. The effect of bepridil was mimicked by 5-(N-4-chlorobenzyl)-2⬘,4⬘-dimethylbenzamil (CBDMB) (IC50⫽6 mol/L), a more selective inhibitor of NCX. In whole-cell patch clamp experiments, bepridil and CB-DMB caused an inward current in spiny neurons recorded from the peri-infarct area but not in the same cells recorded from controls. Interestingly, cholinergic interneurons recorded from the striatal peri-infarct area did not develop an inward current after the application of NCX inhibitors, suggesting that the electrophysiological alterations induced by NCX inhibition are cell-type specific. Bepridil and CB-DMB also induced a suppression of excitatory synaptic currents in most of spiny neurons recorded from the peri-infarct area. This effect was not coupled to a significant change of paired-pulse facilitation suggesting a postsynaptic site of action. Conclusions—Our data indicate that NCX plays a critical role in the maintenance of ionic homeostasis in the peri-infarct area. (Stroke. 2007;38:1614-1620.) Key Words: electrophysiology 䡲 field potential 䡲 ischemia 䡲 Na⫹/Ca2⫹ exchanger 䡲 permanent middle cerebral artery occlusion 䡲 striatum n neuronal cells, the Na⫹/Ca2⫹ exchanger (NCX) plays a fundamental role in controlling Na⫹ and Ca2⫹ homeostasis.1 Depending on the intracellular concentrations of Ca2⫹ and Na⫹, NCX can operate either in the forward mode, coupling the extrusion of Ca2⫹ to the influx of Na⫹ ions, or in the reverse mode, mediating the extrusion of Na⫹ and the influx of the Ca2⫹ ions.1 This feature of NCX is of critical importance considering that changes in the intracellular concentrations of Na⫹ and Ca2⫹ ions are known to occur in pathophysiologic states, such as brain ischemia.2–9 Moreover, a prominent feature of cerebral ischemia is the excessive extracellular accumulation of glutamate and subsequent impairment of intracellular Na⫹ and Ca2⫹ homeostasis, I which results in subsequent cell death.3,10,11 Thus, by controlling intracellular homeostasis of these 2 ions, NCX may play a pivotal role in the events leading to ischemic damage. Reduction of the energy supply leads to a cascade of events including depolarization, influx of Na⫹, and the subsequent reverse operation of the membrane protein that ultimately terminates in intracellular Ca2⫹ overload and irreversible neuronal injury.2,12 Conflicting reports have been published on the role played by NCX during brain ischemia. The inhibition of NCX prevents the activation of phospholipases that occurs after an ischemic insult of the brain.13 Similarly, activation of NCX in its reverse mode plays an important role in cellular Ca2⫹ Received November 24, 2006; accepted December 14, 2006. From Fondazione Santa Lucia IRCCS (A.T., A.Tozzi, B.P., C.C., D.C., G.B., I.B., P.C.), CERC, Rome, Italy; Clinica Neurologica (D.C., G.B., I.B., S.R.), Dip di Neuroscienze, Università di Roma Tor Vergata, Rome, Italy; Division of Pharmacology (A.T., L.A.), Department of Neuroscience, School of Medicine, “Federico II” University of Naples, Naples, Italy; Clinica Neurologica (A.Tozzi, C.C., M.D.F., M.T., P.C.), Dip Specialità MedicoChirurgiche, Università di Perugia, Perugia, Italy. A.T. and B.P. contributed equally to this work and both should be considered first authors. Correspondence to Professor Paolo Calabresi, Clinica Neurologica, Facoltà di Medicina e Chirurgia, Università degli Studi di Perugia, Ospedale Silvestrini, S.Andrea delle Fratte, 06156 Perugia, Italy. E-mail [email protected] © 2007 American Heart Association, Inc. Stroke is available at http://www.strokeaha.org DOI: 10.1161/STROKEAHA.106.478644 1614 Tortiglione et al Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 overload and irreversible damage after energy deprivation.13–16 Conversely, by using in vitro and in vivo models of anoxia and ischemia, it has been demonstrated that the stimulation of NCX activity may help neurons to survive, whereas its pharmacological blockade can compromise their survival.6,7,17,18 Accordingly, we found that the activation of the NCX exerts a protective role during the early phase of oxygen and glucose deprivation in striatal neurons.19 Considering this controversial issue, we have analyzed the effect of the pharmacological inhibition of NCX on the physiological properties of neurons recorded from the striatum, a structure that is highly vulnerable to ischemic insults.4,5,20,21 The mRNA and proteins of the 3 isoforms NCX1, NCX2, and NCX3 are widely expressed in the striatum.9,22 However, their exact location on specific striatal neuronal subpopulations as well as their role in cell-type–specific postischemic striatal damage has been not yet analyzed. Thus, we have recorded the physiological activity of striatal neurons in the peri-infarct area after permanent middle cerebral artery occlusion and their electrical response to the pharmacological inhibition of NCX. In the peri-infarct area, we have also studied the possible differential sensitivity to NCX inhibition of striatal spiny neurons, a neuronal subtype that is highly sensitive to ischemia and energy deprivation, versus striatal cholinergic interneurons, that have been reported to be resistant to these pathological conditions.4,5,20,21 Materials and Methods Animals Sprague-Dawley male rats (250 to 270 grams; Charles River, Lecco, Italy) were used. All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/EEC). Permanent Middle Cerebral Artery Occlusion, Monitoring of Regional Cerebral Blood Flow, and Infarct Size Analysis Methods for permanent middle cerebral artery occlusion have been previously described.23,24 Regional cerebral blood flow was monitored in the ipsilateral parietal cortex to the occluded middle cerebral artery using laser Doppler flowmeter (Periflux System 5000), provided with PC software (PeriSoft PSW 2.0) used to record the blood flow.25 For the staining of infarct size analysis, 2,3,5-triphenyltetrazolium chloride was used as previously described.24 The results are given as infarcted volume, adjusted for edema index, as the percentage of the whole ipsilateral cerebral hemisphere.26 Electrophysiology and Identification of the Peri-Infarct Area Preparation and maintenance of rat corticostriatal slices (270 to 300 m) have been previously described.19,20,27 Slices were kept in artificial cerebrospinal fluid (34° C, O2/CO2) whose composition was (in mmol/L): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3. For synaptic stimulation, bipolar electrodes were used, located in the white matter to activate corticostriatal fibers. Electrophysiological electrodes for extracellular recording (15 to 20 mol/L⍀) were filled with 2 mol/L NaCl. The peri-infarct area was defined through electrophysiological criteria.28 Recording electrodes were initially positioned within the striatum. The stimulating electrode, constantly held 0.3 mm apart, Naⴙ/Ca2ⴙ Exchanger and Ischemia 1615 was progressively placed in up to 8 distinct positions around the recording electrode (360°) to try to elicit extracellular field potentials. We assumed we were in the ischemic core when no reliable field potential (negative deflection from the baseline of at least 0.2 mV) was evocable despite the progressive increase of the stimulation intensity. From the “core” position, the electrodes were progressively (0.5 mm) moved along a single line toward the periphery of the striatum, with the stimulating electrode preceding the recording one. When the field potential appeared, we considered we were in the peri-infarct area. In the “ischemic core” it was not possible to obtain single cell recordings by using whole-cell patch clamp. Conversely, in the peri-infarct area this technique allowed to obtain reliable electrophysiological recordings. This was confirmed analyzing the extension of the ischemic core after 2,3,5-triphenyltetrazolium chloride staining of the slices obtained immediately before and after the one used for the electrophysiological experiments. Whole-Cell Patch Clamp Recordings Whole-cell patch clamp recordings and identification of individual neurons were performed in situ using a differential interference contrast (Nomarski) optical system as previously described.29 Striatal spiny neurons were clamped at ⫺80 mV, whereas striatal aspiny interneurons were clamped at ⫺60 mV, close to their respective resting membrane potentials. All the experiments were performed in the presence of 30 mol/L bicuculline.29 Brain slices were prepared 24 hours after the ischemic insult. Whole patch clamp recordings were obtained within 0.5 to 3 hours after slice preparation. Within this period, the viability of the neurons within the slice was perfect. In fact, although in the ischemic core no recording was possible, in the peri-infarct area as well as in the surrounding normal tissue both spiny neurons and cholinergic interneurons showed intrinsic and synaptic membrane properties similar to those previously reported for “healthy” neurons.27–29 Although from each single slice we could obtain several wholecell recordings, we used only one cell to perform the pharmacological experiments with NCX inhibitors to avoid multiple applications of these drugs on the same brain slice preparation. Statistical Analysis and Drug Application Values represent mean⫾SEM of changes in the respective cell populations. Student t test was used to compare the means. Bepridil (Sigma, Milan, Italy) was applied by dissolving it to the desired final concentration in the saline solution. 5-(N-4-chlorobenzyl)-2⬘,4⬘dimethylbenzamil (CB-DMB) was from Division of Pharmacology, Department of Neuroscience, School of Medicine, “Federico II” University of Naples, Italy. Results Pharmacological Blockade of NCX and Field Potentials Recorded in the Peri-Infarct Area As shown in Figure 1A, the peri-infarct area was detected by morphological and electrophysiological analysis by field potential detections28 at 24 hours after the ischemic insult. From sham-operated animals, slices were obtained at the same time window. Application of bepridil (100 mol/L) in the extracellular medium for 15 minutes significantly reduced the amplitude of the field potential obtained 24 hours (n⫽8, ***P⬍0.0001 field potential amplitude t⫽⫺15 versus t⫽40) after the permanent middle cerebral artery occlusion (Figure 1B and 1C). No significant inhibition was detected in slices prepared from sham-operated animals at the same timewindow (100 mol/L, n⫽8, P⬎0.05 field potential amplitude t⫽⫺15 versus t⫽40; Figure 1B and 1C). As shown in Figure 2, the inhibitory action of bepridil on the amplitude of field potentials recorded from the periinfarct area of rats exposed to permanent middle cerebral 1616 Stroke May 2007 Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 Figure 1. The inhibition of the NCX induced by bepridil reduces the field potential amplitude recorded from the peri-infarct area in rat corticostriatal slices. A, Electrophysiological and morphological characterization of the peri-infarct area (P) and of the ischemic core (C). B, The plot represents the effect of bath application of 100 mol/L bepridil in corticostriatal slices obtained at 24 hours after permanent middle cerebral artery occlusion in multiple experiments and in slices obtained from sham-operated animals. C, Traces represent examples of corticostriatal field potentials recorded before, during, and after bath application of 100 mol/L bepridil in slices obtained from sham-operated animals (upper row) and from animals exposed to focal ischemia (lower rows). Figure 2. Bepridil decreases the field potential amplitude in ischemic slices in a dose-dependent manner (A). The selective NCX inhibitor CB-DMB mimics the inhibitory effects of bepridil of field potential amplitude in a dose-dependent manner (B). The graph shows the dose-response curve obtained from ischemic tissue and from sham-operated striata. Each data point represents at least 3 single experiments. Traces were obtained from single experiments in ischemic and sham-operated animals (C). artery occlusion was dose-dependent (n⫽6 for each dose; IC50⫽68 mol/L). The inhibitory action of bepridil was also reproduced with CB-DMB as shown in the dose-response curve of Figure 2 (IC50 of 6 mol/L). This compound, synthesized by the amiloride discoverer Cragoe,30 is the most potent and selective pyrazine derivative, which inhibits forward and reverse mode of NCX operation.31–36 Interestingly, this NCX inhibitor, at the incubation time intervals used in the present study, does not interfere with L-type Ca2⫹ channels.37 However, more recent putative NCX inhibitors including KB-R7943 Tortiglione et al Naⴙ/Ca2ⴙ Exchanger and Ischemia 1617 Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 Figure 3. The inhibition of the NCX produces an inward current in striatal medium spiny neurons (MS), but not in large cholinergic interneurons (LA) recorded from the peri-infarct area. A, Changes of the membrane currents detected by wholecell patch clamp recordings in spiny neurons versus cholinergic interneurons in slices obtained either from sham-operated animals (upper rows) or from animals exposed to focal ischemia (lower rows). The holding potential was kept constant during all the experiment and it was ⫺80 mV for spiny neurons and ⫺60 mV for cholinergic interneurons. B, The histogram shows the percentage of spiny neurons showing irreversible inward currents in sham-operated rats and in animals exposed to ischemia 24 hours before. C, The plot shows the time-course of the bepridil-induced inward currents in spiny neurons recorded from the peri-infarct area. D, The plot shows an example of a current–voltage relationship measured from spiny neurons recorded in the periinfarct area and obtained by delivering both positive and negative voltage steps from the holding potential. and SN-6 can also interfere with several ion transporters and channels.38 To rule out that the reduction of the field potential observed in the peri-infarct area after the administration of NCX inhibitors could be caused by tissue deterioration during electrophysiological recordings rather than by the specific drug effect, we performed field potential recordings in the peri-infarct area also in control medium. Under this experimental condition, we have observed no change of the field potential amplitude throughout the recording time (n⫽9, data not shown). Effect of NCX Inhibitors on Membrane Currents of Medium Spiny Neurons and Large Aspiny Interneurons Recorded in the Peri-Infarct Area To asses the possible mechanism underlying the irreversible loss of the field potential amplitude caused by bepridil in spiny neurons after ischemia, we measured, by using wholecell patch clamp recordings, the effects of NCX inhibitors on somatic currents recorded either in the striatal peri-infarct area or in the striatum of sham-operated rats. As shown in Figure 3A through 3C, 100 mol/L bepridil induced an inward current in most of the medium spiny neurons (8/12) recorded from the peri-infarct area of striatal slices obtained 24 hours after the ischemic insult (P⬍0.01 15 minutes after the application of bepridil). Conversely, spiny neurons recorded from sham-operated striata did not show significant changes of resting membrane currents (11/11) (P⬎0.05, 15 minutes after the application of bepridil). Interestingly, cholinergic interneurons did not show bepridil-induced current when recorded either from the peri-infarct area (7/7) (P⬎0.05, 15 minutes after the application of bepridil), or from control tissue (6/6) (P⬎0.05, 15 minutes after the application of bepridil). The bepridil-induced inward current detected in the spiny neurons from the peri-infarct area developed within 5 minutes from the onset of the application of bepridil and reached a peak at ⬇15 minutes. This inward current did not tend to reverse after bepridil removal. Figure 3D shows a voltage clamp experiment performed by applying increasing voltage steps in both directions to detect a reversal potential for the bepridil-induced inward current. The reversal potential for this current was estimated close to ⫺45 mV (n⫽4). CB-DMB 10 mol/L mimicked the effects of bepridil, inducing an inward current (at least 50 pA) in 5 of 6 spiny neurons recorded in the peri-infarct area (data not shown). No current was detected in 5 spiny neurons obtained from sham-operated rats. Moreover, no current was induced in cholinergic interneurons recorded either from ischemic tissue (n⫽4) or from sham-operated animals (n⫽5) (data not shown). Effect of NCX Inhibitors on Evoked Excitatory Synaptic Current Recorded From Medium Spiny Neurons in the Peri-Infarct Area Figure 4 shows that application of bepridil causes a progressive reduction of the amplitude of excitatory postsynaptic currents (EPSCs) recorded from medium spiny neurons in the peri-infarct area (n⫽7, P⬍0.05). Also in this case, as well as for the bepridil-induced inward current, the EPSCs did not recover after the interruption of the drug application. In medium spiny neurons obtained from the striatum of shamoperated rats, no change of EPSC amplitude was observed (n⫽7, P⬎0.05). To investigate whether the depression of EPSCs induced by bepridil in spiny neurons from the peri-infarct area was dependent on presynaptic or postsynaptic sites of action, we measured synaptic responses to a pair of stimuli before and during the application of the NCX inhibitor. In these experiments, interstimulus interval was 60 ms. Paired-pulse modification of neurotransmission has been studied extensively and is attributed to a presynaptic change in release probabil- 1618 Stroke May 2007 Figure 4. Inhibition of the NCX by bepridil decreases excitatory synaptic currents measured in spiny neurons recorded from the peri-infarct area. A, The plot shows the time course of the EPSC amplitude detected in spiny neurons obtained either from sham-operated animals or from rats exposed to ischemia. The traces in the lower part of the panel show examples of EPSCs recorded before and after the application of bepridil in these 2 experimental conditions. In both the experiments the holding potential was ⫺80 mV. B, The histogram represent the EPSC2/EPSC1 ratio in a paired-pulse experiment before and after the application of 100 mol/L bepridil. Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 ity. An increase in the ratio of the second pulse response to the first pulse response (EPSC2/EPSC1) indicates a decrease in the release probability. The depression of the EPSC amplitude induced by 100 mol/L bepridil was not associated with a significant increase in this ratio (n⫽5, P⬎0.05; Figure 4B), suggesting that postsynaptic mechanisms play a role in the observed effect. The synaptic effects of bepridil were mimicked by 10 mol/L CB-DMB (n⫽7, data not shown). Discussion The present study represents the first electrophysiological demonstration that the activation of NCX maintains ionic homeostasis in the peri-infarct area after focal ischemia. We found, in fact, that the pharmacological inhibition of NCX by bepridil and CB-DMB causes an irreversible loss of field potential amplitude recorded from the striatal area surrounding the ischemic core. These effects were dose-related and irreversible suggesting that inhibition of NCX can trigger a cascade of detrimental events leading to neuronal damage. The hypothesis that the electrophysiological action of bepridil and CB-DMB are closely linked to the ischemia-triggered events is also confirmed by the evidence that these drugs failed to alter the field potential amplitude in slices obtained from sham-operated rats. Bepridil is not considered a selective NCX inhibitor. In fact, this drug inhibits sodium and calcium channels and modulates potassium conductances. Nevertheless, the use of a more specific NCX inhibitor, such as CB-DMB,18,31,33,34 mimicking the electrophysiological effects of bepridil strongly support the hypothesis that the effects we observed are dependent on the selective inhibition of NCX. The change of the expression in NCX1, NCX2, and NCX3 gene products after permanent middle cerebral artery occlusion seems to suggest that these isoforms can be involved in the process leading to cell death or survival in those areas that are interested by the ischemic insult.6,7,9 According to our electrophysiological and pharmacological data, it has been shown that after knocking down 2 of these proteins, NCX1 and NCX3, a remarkable enlargement of the infarct volume occurred. Such a phenomenon was associated with a worsening of neurological deficits.7 Accordingly, the pharmacological inhibition of NCX leads to an enlargement of the infarct size.6 By using single-cell patch clamp recordings, we have demonstrated that the pharmacological blockade of NCX produced an inward current in spiny neurons recorded in the peri-infarct area, whereas no current was detected in cells obtained from sham-operated animals. The estimated reversal potential for this inward current was ⬇⫺45 mV, indicating that multiple events rather than a single conductance seem to be involved.27 The single cell experiments gave us also the possibility to demonstrate that the pharmacological action of bepridil and CB-DMB were cell-type–specific. In fact, no inward current was detected from striatal large cholinergic interneurons recorded from the peri-infarct area. Noticeably, striatal spiny neurons are vulnerable to energy deprivation,4,20,21,29 whereas cholinergic interneurons have been reported to be resistant to ischemia.4,5,20,21 Thus, we can argue that the different expression of NCX among various striatal neuronal subtypes might take part in the mechanisms controlling cell-type–specific vulnerability to ischemia within the striatum and, possibly, in other brain areas. However, as an alternative hypothesis, we can consider the possibility that although the expression of NCX is similar in spiny neurons and cholinergic interneurons, these 2 neuronal subtypes respond in a distinct manner to NCX inhibition because they have a differential capability to compensate alterations of ion concentrations. The inward current recorded from spiny neurons during application of NCX inhibitors was paralleled by a progressive Tortiglione et al Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 decrease of the amplitude of the evoked EPSCs. This latter effect may also represent the synaptic event underlying loss of the field potential amplitude detected by extracellular recordings. In fact, the field potential amplitude reflects the synchronous discharge of several neurons adjacent to the recording electrode and activated by excitatory fibers. Interestingly, the reduction of the EPSC amplitude was not coupled to changes in EPSC2/EPSC1 ratio, suggesting that postsynaptic rather than presynaptic mechanisms seem to underlie this event. An interesting issue that should be considered in future studies is related to the potential recovery of the peri-infarct area and its relationship with the functional activity of NCX at different time windows after the ischemic insult. The 3 isoforms of NCX that are present in the striatum are constitutively expressed not only in neurons but also in glia.39 Interestingly, the expression of this antiporter can be altered under particular conditions, such as hypoxia.9 Cerebral edema is known to exacerbate neuronal damage induced by ischemia. Accordingly, it has been recently reported that a nonselective cation channel is expressed after brain ischemia and is crucially involved in development of cerebral edema.40 However, no evidence has never been provided on an involvement of NCX on water distribution either in normal tissue or in focal ischemia. NCX is also expressed in peripheral cells such as macrophages and leukocytes1 migrating in the brain after the ischemic insult; therefore, a hypothetical role played by the modification of NCX in these cells cannot be excluded. Nevertheless, the changes observed in the electrophysiological behavior of spiny and cholinergic interneurons seem to favor the idea that the action of the NCX inhibitors is mainly directed to neuronal cells rather than in peripheral cells. In conclusion, our data suggest that in the peri-infarct area, NCX plays a role in maintaining ionic homostasis because its pharmacological block leads to detrimental effects after focal cerebral ischemia. Moreover, this effect is selectively expressed in neurons particularly vulnerable to energy deprivation. Acknowledgments We thank Mr Cristiano Spaccatini and Mr Massimo Tolu for their excellent technical support. Sources of Funding This work was supported by grants from Ministero della Salute (Ricerca Finalizzata 2004, 2005 IRCCS to P.C. and B.P.), FIRB 2001, Fondazione Cassa Risparmio Perugia (to P.C.). Disclosures None. References 1. Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763– 854. 2. Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. 3. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79: 1431–1568. 4. Calabresi P, Centonze D, Bernardi G. Cellular factors controlling neuronal vulnerability in the brain: a lesson from the striatum. Neurology. 2000;55:1249 –1255. Naⴙ/Ca2ⴙ Exchanger and Ischemia 1619 5. Centonze D, Marfia GA, Pisani A, Picconi B, Giacomini P, Bernardi G, Calabresi P. Ionic mechanisms underlying differential vulnerability to ischemia in striatal neurons. Prog Neurobiol. 2001;63:687– 696. 6. Pignataro G, Tortiglione A, Scorziello A, Giaccio L, Secondo A, Severino B, Santagada V, Caliendo G, Amoroso S, Di Renzo G, Annunziato L. Evidence for a protective role played by the Na⫹/Ca2⫹ exchanger in cerebral ischemia induced by middle cerebral artery occlusion in male rats. Neuropharmacology. 2004;46:439 – 448. 7. Pignataro G, Gala R, Cuomo O, Tortiglione A, Giaccio L, Castaldo P, Sirabella R, Matrone C, Canitano A, Amoroso S, Di Renzo G, Annunziato L. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke. 2004;35:2566 –2570. 8. Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na⫹/Ca2⫹ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633– 654. 9. Boscia F, Gala R, Pignataro G, de Bartolomeis A, Cicale M, AmbesiImpiombato A, Di Renzo G, Annunziato L. Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na⫹/Ca2⫹ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J Cereb Blood Flow Metab. 2006;26:502–517. 10. Kiedrowski L, Brooker G, Costa E, Wroblewski JT. Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron. 1994;12:295–300. 11. Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239 –247. 12. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399 – 415. 13. Pilitsis JG, Diaz FG, O’Regan MH, Phillis JW. Inhibition of Na(⫹)/Ca(2⫹) exchange by kb-r7943, a novel selective antagonist, attenuates phosphoethanolamine and free fatty acid efflux in rat cerebral cortex during ischemia-reperfusion injury. Brain Res. 2001;916:192–198. 14. Schroder UH, Breder J, Sabelhaus CF, Reymann KG. The novel Na⫹/Ca2⫹ exchange inhibitor KB-R7943 protects ca1 neurons in rat hippocampal slices against hypoxic/hypoglycemic injury. Neuropharmacology. 1999;38:319 –321. 15. Breder J, Sabelhaus CF, Opitz T, Reymann KG, Schroder UH. Inhibition of different pathways influencing Na(⫹) homeostasis protects organotypic hippocampal slice cultures from hypoxic/hypoglycemic injury. Neuropharmacology. 2000;39:1779 –1787. 16. Li S, Jiang Q, Stys PK. Important role of reverse Na(⫹)-Ca(2⫹) exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84:1116 –1119. 17. Amoroso S, Sensi S, di Renzo G, Annunziato L. Inhibition of the Na(⫹)-Ca2⫹ exchanger enhances anoxia and glucopenia-induced [3H]aspartate release in hippocampal slices. J Pharmacol Exp Ther. 1993;264: 515–520. 18. Amoroso S, Tortiglione A, Secondo A, Catalano A, Montagnani S, Di Renzo G, Annunziato L. Sodium nitroprusside prevents chemical hypoxia-induced cell death through iron ions stimulating the activity of the Na⫹-Ca2⫹ exchanger in C6 glioma cells. J Neurochem. 2000;74: 1505–1513. 19. Calabresi P, Marfia GA, Amoroso S, Pisani A, Bernardi G. Pharmacological inhibition of the Na(⫹)/Ca(2⫹) exchanger enhances depolarizations induced by oxygen/glucose deprivation but not responses to excitatory amino acids in rat striatal neurons. Stroke. 1999;30:1687–1694. 20. Calabresi P, Saulle E, Centonze D, Pisani A, Marfia GA, Bernardi G. Post-ischaemic long-term synaptic potentiation in the striatum: a putative mechanism for cell type-specific vulnerability. Brain. 2002; 125:844 – 860. 21. Calabresi P, Centonze D, Pisani A, Cupini L, Bernardi G. Synaptic plasticity in the ischaemic brain. Lancet Neurol. 2003;2:622– 629. 22. Papa M, Canitano A, Boscia F, Castaldo P, Sellitti S, Porzig H, Taglialatela M, Annunziato L. Differential expression of the Na⫹-Ca2⫹ exchanger transcripts and proteins in rat brain regions. J Comp Neurol. 2003;461: 31– 48. 23. Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1:53– 60. 24. Tortiglione A, Pignataro G, Minale M, Secondo A, Scorziello A, Di Renzo GF, Amoroso S, Caliendo G, Santagada V, Annunziato L. Na⫹/Ca2⫹ exchanger in Na⫹ efflux-Ca2⫹ influx mode of operation exerts a neuroprotective role in cellular models of in vitro anoxia and in vivo cerebral ischemia. Ann N Y Acad Sci. 2002;976:408 – 412. 1620 Stroke May 2007 Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 25. Otori T, Greenberg JH, Welsh FA. Cortical spreading depression causes a long-lasting decrease in cerebral blood flow and induces tolerance to permanent focal ischemia in rat brain. J Cereb Blood Flow Metab. 2003;23:43–50. 26. Reglodi D, Somogyvari-Vigh A, Vigh S, Kozicz T, Arimura A. Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke. 2000;31:1411–1417. 27. Calabresi P, Marfia GA, Centonze D, Pisani A, Bernardi G. Sodium influx plays a major role in the membrane depolarization induced by oxygen and glucose deprivation in rat striatal spiny neurons. Stroke. 1999;30:171–179. 28. Picconi B, Tortiglione A, Barone I, Centonze D, Gardoni F, Gubellini P, Bonsi P, Pisani A, Bernardi G, Di Luca M, Calabresi P. NR2b subunit exerts a critical role in postischemic synaptic plasticity. Stroke. 2006;37: 1895–1901. 29. Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, Pavon N, Pisani A, Bernardi G, Moratalla R, Calabresi P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23:8506 – 8512. 30. Kleyman TR, Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol. 1988;105:1–21. 31. Andreeva N, Khodorov B, Stelmashook E, Cragoe E Jr, Victorov I. Inhibition of Na⫹/Ca2⫹ exchange enhances delayed neuronal death elicited by glutamate in cerebellar granule cell cultures. Brain Res. 1991; 548:322–325. 32. Wettwer E, Himmel H, Ravens U. Amiloride derivatives as blockers of NA⫹/CA2⫹ exchange: effects on mechanical and electrical function of guinea-pig myocardium. Pharmacol Toxicol. 1992;71:95–102. 33. Wacholtz MC, Cragoe EJ Jr, Lipsky PE. Delineation of the role of a Na⫹/Ca2⫹ exchanger in regulating intracellular Ca2⫹⫹ in t cells. Cell Immunol. 1993;147:95–109. 34. Amoroso S, De Maio M, Russo GM, Catalano A, Bassi A, Montagnani S, Renzo GD, Annunziato L. Pharmacological evidence that the activation of the Na(⫹)-Ca2⫹ exchanger protects C6 glioma cells during chemical hypoxia. Br J Pharmacol. 1997;121:303–309. 35. Li Y, Woo V, Bose R. Platelet hyperactivity and abnormal Ca(2⫹) homeostasis in diabetes mellitus. Am J Physiol Heart Circ Physiol. 2001;280:H1480 –H1489. 36. Roberts DE, McNicol A, Bose R. Mechanism of collagen activation in human platelets. J Biol Chem. 2004;279:19421–19430. 37. Sharikabad MN, Cragoe EJ Jr, Brors O. Inhibition by 5-N-(4chlorobenzyl)-2⬘,4⬘-dimethylbenzamil of Na⫹/Ca2⫹ exchange and l-type Ca2⫹ channels in isolated cardiomyocytes. Pharmacol Toxicol. 1997;80: 57– 61. 38. Iwamoto T, Inoue Y, Ito K, Sakaue T, Kita S, Katsuragi T. The exchanger inhibitory peptide region-dependent inhibition of Na⫹/Ca2⫹ exchange by SN-6 [2-[4-(4-nitrobenzyloxy)benzyl]thiazolidine-4-carboxylic acid ethyl ester], a novel benzyloxyphenyl derivative. Mol Pharmacol. 2004;66: 45–55. 39. Nagano T, Kawasaki Y, Baba A, Takemura M, Matsuda T. Up-regulation of Na(⫹)-Ca2⫹ exchange activity by interferon-gamma in cultured rat microglia. J Neurochem. 2004;90:784 –791. 40. Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed sur1-regulated Nc(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433– 440. Na+/Ca2+ Exchanger Maintains Ionic Homeostasis in the Peri-Infarct Area Anna Tortiglione, Barbara Picconi, Ilaria Barone, Diego Centonze, Silvia Rossi, Cinzia Costa, Massimiliano Di Filippo, Alessandro Tozzi, Michela Tantucci, Giorgio Bernardi, Lucio Annunziato and Paolo Calabresi Downloaded from http://stroke.ahajournals.org/ by guest on June 15, 2017 Stroke. 2007;38:1614-1620; originally published online March 29, 2007; doi: 10.1161/STROKEAHA.106.478644 Stroke is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 2007 American Heart Association, Inc. All rights reserved. Print ISSN: 0039-2499. Online ISSN: 1524-4628 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://stroke.ahajournals.org/content/38/5/1614 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Stroke can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in the Permissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Stroke is online at: http://stroke.ahajournals.org//subscriptions/