Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
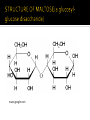
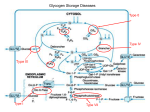
WHAT IS GLYCOGEN STORAGE DISEASES (GSD) ? - GSD has 2 classes of cause : (a) Genetic (b) Acquired -Caused by a genetic enzyme defect that is inherited from both parents. -Normally, enzymes help convert glucose into glycogen for storage and other enzymes convert the glycogen back to glucose when quick energy is needed, like during exercises. In a person with a GSD, some of these enzymes are defective. This causes the build up of abnormal and types of glycogen in liver and/or muscle tissues. In livestock , acquired GSD is caused by intoxication with the alkaloid castanospermine. Low blood sugar. Enlarged liver. Slow growth. Muscle cramps. Also known as ‘’VON – GIERKE’S DISEASE’’. GSD Type 1 is the most common of the GSD and accounts for 90% of all GSD cases. It is results from deficiency of the enzyme glucose-6-phosphatase. This deficiency impairs the ability of the liver to produce glucose from glycogen. Since these are the two principal metabolic mechanisms by which the liver supplies glucose to the rest of the body during periods of fasting, it causes severe hypoglycemia ( deficiency of glucose in bloodstream). Reduced glycogen breakdown results in increased glycogen storage in liver and kidneys, causing enlargement of both. Enlarged liver and kidneys. Low blood sugar. High levels of uric acid in the blood. Impaired growth and delayed puberty. Bone thinning from osteoporosis. Increased mouth ulcers and infection. Frequent or continuous feedings of cornstarch or other carbohydrates are the principal treatment. GLYCOGEN STORAGE DISEASES (GSD) TYPE II ? Pompe’s disease also referred to as Glycogen Storage Disease Type II or acid maltase deficiency, is an autosomal recessive disorder of glycogen metabolism caused by a deficiency of the lysosomal enzyme acid glucosidase. People affected with this disease are unable to degrade glycogen stored in the lysosome and thus leading to the accumulation of glycogen in lysosomal storage vacuoles. Pompe’s disease has been divided into three forms defined by age of onset and progression of symptoms. The three forms include infantile onset, juvenile onset and adult onset. In the infantile form of the disease, patients display cardiac impairment, which is fatal before two years of life. Patients with juvenile or adult forms can present diaphragm involvement leading to respiratory failure. The adult onset symptoms involve generalized muscle weakness and wasting of respiratory muscles in the trunk, lower limbs, and diaphragm. Glycogen is mostly found in the liver and skeletal muscles. The polymer is composed of units of glucose linked α-1-4 with branches occurring at a-16, approximately every 8-12 residues. The end of the molecule containing a free carbon number one on glucose is called a reducing end. The other ends are all called non-reducing ends http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=stryer.figgrp.2912 Glycogen molecules are very large in size Therefore inability to degrade them results to a large accumulation of normal structure in the lysosomes of all cells. The excess storage of glycogen in the vacuoles is the consequence of defects in the lysosomal hydrolase. The acid a-glucosidase normally designated as GAA gene. Glycogen storage disease type II has been shown to be caused by missense, nonsense and splice-site mutations, partial deletions and insertions. Some mutations are specific to certain ethnic groups. The normal function of acid a-glucosidase is to hydrolyze both a-1,4- and a-1,6glucosidic linkages at acid Ph. The activity of the enzyme leads to the complete hydrolysis of glycogen which is its natural substrate. As would be expected from this activity, deficiency in acid a-glucosidase leads to the accumulation of structurally normal glycogen in numerous tissues, most notably in cardiac and skeletal muscle. www.google.com The lysosomal α-1,4-glucosidase was found to be active at pH 4. However, its activity is not present in the liver, heart and skeletal muscles of children with pompe’s disease. Although the lysosomal α-1,4-glucosidase is often referred to as maltase, the enzyme is known to have a broader specificity in that it acts also on the outer chains of glycogen. www.google.com Symptoms Type II, usually found in Lapland dogs, is characterized by vomiting, progressive muscle weakness, and cardiac abnormalities. Death usually occurs before two years of age. Causes Type II from a deficiency of acid glucosidase. Other animals Acid maltase-deficient Japanese quails Australian cattle GLYCOGEN STORAGE DISEASES (GSD) TYPE III ? known as Forbes-Cori disease or limit dextrinosis Due to inability to produce enough glycogen debranching enzyme ( GDE ) The gene responsible for making (GDE) (Glucosyl Transferase & ɑ-1,6-Glucosidae) is the AGL gene Normal Glycogen Debranching First, phosphorylase removes 7 glucose molecules (7 black circles from the unbranched outer part of glycogen molecule) from the other glucose molecules in a glycogen molecule until only 4 glucosyl units (3 green circles and the 1 red circle) remain before an alpha1,6 branch point . The transferase debranching enzyme then transfers the 3 (green) glucose residues from the short branch to the end of an adjacent branch of the glycogen molecule The glucosidase debranching enzyme then removes the glucose molecule(the red circle) remaining at the alpha1,6 branch point. Abnormal Glycogen Debranching Mutation of AGL gene = reduce GDE production = GSD III = Incomplete debranching of glycogen = Less glucose released , Glycogen stored as limit dextrin structure If one copy of the AGL gene is altered but the second copy is not, then the body can follow the instructions on the second copy in order to produce enough debranching enzyme. When both copies of an individual’s AGL gene are altered, the body is unable to read any instructions on how to make the proper amount of debranching enzyme. As a result, the individual has GSD III. GSD IIIa - includes muscle and liver involvement GSD IIIb - liver involvement but no muscle involvement GSDIIIc (extremely rare) - Presumably the result of deficiency of only glucosidase debranching activity GSDIIId (extremely rare)- Presumably the result of deficiency of only transferase debranching activity GLYCOGEN STORAGE DISEASES (GSD) IV and V? GSD type IV, also known as amylopectinosis or Andersen disease, is a rare disease that leads to early death. In 1956, Andersen reported the first patient with progressive hepatosplenomegaly and accumulation of abnormal polysaccharides. The main clinical features are liver insufficiency and abnormalities of the heart and nervous system. Type IV, or Andersen's disease, is caused by glycogen branches enzyme deficiency in the liver, brain, heart, skeletal muscles, and skin fibroblasts. The glycogen constructed in GSD IV is abnormal and insoluble. As it accumulates in the cells, cell death leads to organ damage. Infants born with GSD IV appear normal at birth, but are diagnosed with enlarged livers and failure to thrive within their first year. Infants who survive beyond their first birthday develop cirrhosis of the liver by age 3-5 and die as a result of chronic liver failure. Symptoms and sings: 1.The liver (cirrhosis) progressive and spleen enlarge. 2. Death typically occurs by five years of age. GSD type V, also known as McArdle disease, affects the skeletal muscles. Initial signs of the disease usually develop in adolescents or adults. Muscle phosphorylase deficiency adversely affecting the glycolytic pathway in skeletal musculature causes GSD type V. Like other forms of GSD, McArdle disease is heterogeneous. Caused by glycogen phosphorylase deficiency in skeletal muscles. The phosphorylase enzyme plays a vital role in the breakdown of glycogen into glucose. glucose can not be released from the glycogen stored in skeletal muscles to create energy. People with type V GSD experience problems performing and completing most exercises, especially anaerobic exercises. muscle weakness and cramping brought on by exercise, as well as burgundy-colored urine after exercise due to myoglobin (a breakdown product of muscle) in the urine.