Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
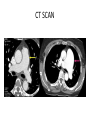
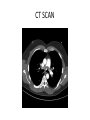
PULMONARY HYPERTENSION ADIL K. WARSY M.D. PGYV PULMONARY FELLOW FINANCIAL DISCLOSURES • None OBJECTIVES • • • • • • • • • Learn to define the disorder Classification and prevalence Pathophysiology and histopathology Presentation Screening and diagnostic methods Functional classification Treatment approaches Prognosis PAH and pregnancy DEFINITIONS • PULMONARY ARTERIAL HYPERTENSION Syndrome resulting from restricted blood flow through the pulmonary arterial circulation from vascular proliferation, aberrant remodeling and in situ thrombosis resulting in increased pulmonary vascular resistance and ultimately right sided heart failure. • Hemodynamic state characterized by - Sustained elevation of mean pulmonary artery pressure to >25mm Hg at rest or >30mm Hg with exercise -Mean PCWP and LVEDP<15mm Hg PULMONARY AND SYSTEMIC CIRCULATION • Standard pressures -Mean pulmonary artery pressure, 14mm Hg -PCWP, 8-12 mm Hg -LVEDP, 6-12 mm Hg -Systemic arterial pressure <120/80 mm Hg • PAH pressures -Mean pulmonary artery pressure > 25mm Hg -PCWP and LVEDP < 15 mm Hg -Systemic arterial pressure < 120/80 mm Hg INCIDENCE/PREVALENCE • PAH is rare • Estimated prevalence of 3050 cases per million • Most common in young women • Mean age of diagnosis 36 years • The prevalence in certain at-risk groups is higher • HIV-infected patients (0.50%) • Sickle cell disease (2040%) • Systemic sclerosis (16%)5 • True prevalence may be higher CLASSIFICATION • OLD CLASSIFICATION Primary and Secondary • In 1998 it was proposed to classify based on -Disorders in between the ones affecting the arterial part of pulmonary circulation -Disorders affecting venous part of the circulation -Disorders affecting the circulation by altered respiratory function. CLASSIFICATION • Third World Conference on Pulmonary Hypertension, held in Venice, Italy, in 2003. The major changes noted in these revisions were as follows: (1) Replacement of the term primary PH with -Idiopathic pulmonary arterial hypertension (IPAH) -Familial pulmonary arterial hypertension (FPAH) when supported by genetic testing, (2) Abandonment of the term secondary PH WHO CLASSIFICATION • Group I. Pulmonary arterial hypertension (PAH) 1. 2. 3. – – – – – – 4. – – 5. Idiopathic (IPAH) Familial (FPAH) Associated with (APAH): Connective tissue disease Congenital systemic-to-pulmonary shunts-Eisenmenger’s syndrome Portal hypertension HIV infection Drugs and toxins Other (thyroid disorders, glycogen storage disease, Gaucher's disease, hereditary haemorrhagic telangiectasia, haemoglobinopathies, myeloproliferative disorders, splenectomy Associated with significant venous or capillary involvement Pulmonary veno-occlusive disease (PVOD) Pulmonary capillary haemangiomatosis (PCH) Persistent pulmonary hypertension of the newborn (PPHN) WHO CLASSIFICATION • Group II. Pulmonary hypertension associated with left heart diseases • Group III. Pulmonary hypertension associated with respiratory diseases and / or hypoxemia (including COPD) • Group IV. Pulmonary hypertension due to chronic thrombotic and/or embolic disease -Thromboembolic obstruction of proximal pulmonary arteries -Thromboembolic obstruction of distal pulmonary arteries -Pulmonary embolism (tumor, parasites, foreign material) -Sickle cell disease • Group V. Miscellaneous group eg. sarcoidosis, histiocytosis X and lymphangiomatosis WHO GROUP I • IPAH (idiopathic pulmonary arterial hypertension - Leading cause almost 40% of the cases - No risk factor, family history or genetic mutation. - More common in women - Mean age 52 WHO GROUP I • FPAH (Familial pulmonary arterial hypertension) - At least 100 families have been identified world wide - Accounts for >6% of all cases of PAH - Pedigree analysis shows an autosomal dominant inheritance with reduced penetrance ( 10% to 20% of carriers are affected) - Two genes in TGF-B receptor family have been linked to FPAH - BMPR-2 and ALK-1 - Mutations in bone morphogenetic protein receptor type 2 (BMPR2) have been detected in approx. 71% of patients with FPAH - Mutations in BMPR2 have been detected in 26% of IPAH cases PATHOPHYSIOLOGY • Exact cause of PAH remains unknown • Endothelial dysfunction occurs early on in the disease process • Endothelial dysfunction results in • Reduced production of vasodilators • Over production of vasoconstrictors • Endothelial and smooth muscle cell proliferation • Remodelling of the pulmonary vascular bed and increased pulmonary vascular resistance (PVR) • Increase in pulmonary vascular resistance (PVR) • Leads to right ventricular overload • Leads to right ventricular failure and premature death PATHOPHYSIOLOGY • Three pathways are involved: 1. Prostacyclin (prostaglandin) 2. Nitric oxide (NO)− Cyclic guanosine monophosphate ( GMP)- phosphodiesterase 5 3. Endothelin • Platelets likely play an important role as pro-coagulants by increasing the platelet release of serotonin, vascular endothelial growth factor, and platelet-derived growth factor. PATHOPHYSIOLOGY • Reduced production of vasodilators • Prostacyclin • potent vasodilator • potent inhibitor of platelet activation • therapy with synthetic forms of prostacyclin may help to correct this deficiency • Nitric oxide • potent vasodilator • possesses anti-proliferative properties • vasodilatory effect is mediated by cGMP • rapidly degraded by phosphodiesterases PATHOPHYSIOLOGY • Increased production of vasoactive compounds • Endothelin (ET) • elevated levels are seen in PAH patients • levels correlate with disease severity • deleterious effects mediated through ETA and ETB receptors5 • • • • fibrosis hypertrophy and cell proliferation inflammation vasoconstriction • Endothelin receptor antagonists can block these effects • ET, nitric oxide and Prostacyclin have been the principal focus of research into treatments for PAH DISEASE PATHWAY IN PAH HISTOPATHOLOGY PRESENTATION SYMPTOMS • High resistance to blood flow through the lungs causes right heart dysfunction and produces: • • • • • • • • • • Dyspnea Fatigue Dizziness Syncope Peripheral edema Anginal chest pain, particularly during physical exercise Hemoptysis Hoarseness Anorexia RUQ discomfort • Symptoms are non-specific and are commonly attributed to other conditions • Over time, symptoms become more severe and limit normal daily activities PRESENTATION • • • • • • • CLINICAL SIGNS Jugular venous distension Prominent P2 Right ventricle heave Tricuspid insufficiency murmur Right side S3 heart sound Hepatomegaly Peripheral edema WHY IT IS A DIAGNOSTIC DILEMMA? • Low awareness/not a condition seen every day by general physicians • It often comes in disguise: • Non-specific symptoms that are often mild and common to other conditions • Confusion with other diseases such as asthma, COPD and other cardiovascular disorders • No simple means of excluding PAH STEPWISE APPROACH TO DIAGNOSE Diagnosis requires a series of investigations in a four-stage approach: • Clinical suspicion of pulmonary hypertension Symptoms, screening and incidental findings • Detection of pulmonary hypertension ECG, chest radiograph, Doppler echocardiography • Identification of other causes of pulmonary HTN Pulmonary function test, blood gas samples, HRCT • PAH evaluation and classification Functional capacity and hemodynamics SCREENING HIGH RISK POPULATION • Key to early diagnosis – screening high risk populations: – Family members of a patient with familial Pulmonary Arterial Hypertension (FPAH) – Patients with systemic sclerosis (SSc) – Patients with HIV – Patients with portopulmonary hypertension (PoPH) • International guidelines recommend annual screening with Doppler echocardiography. • Right heart catheterisation still only method for definitive diagnosis 1-3 TOOLS FOR SCREENING • Clinical suspicion • History and Physical • Doppler echocardiography. SCREENING HIGH RISK POPULATION Results of a disease registry in France: Without screening, the majority of patients were diagnosed in WHO FC III or FC IV, and only 24% of patients were in WHO FC II at diagnosis. ECG ECHOCARDIOGRAPHY Specificity moderate : PAP overestimated in patients with advanced lung disease NONINVASIVE TESTING • Full PFTs with DLCO help identify COPD Interstitial lung disease • ABG • CT scan Lung pathology Pulmonary embolism • HRCT Interstitial lung disease CT SCAN CT SCAN VQ SCAN in CTEPH CLARIFY ETIOLOGY • Patients with PAH on echocardiogram H/O EDS, snoring, apneas Obtain NPSG Arthralgias, arthritis, myalgias, skin lesions Obtain serology for CTD Risk of/ exposure to HIV Screen for CTEPH Many patients will not have h/o VTE VQ scan should be ordered. RIGHT HEART CATHETERISATION-THE GOLD STANDARD RIGHT HEART CATHETERISATION-THE GOLD STANDARD • RHC should always assess • • • • • • right atrial pressure (RAP) systolic, diastolic and mean pulmonary arterial pressure (PAP) pulmonary capillary wedge pressure (PCWP) cardiac output / index PVR and systemic vascular resistance blood pressure and arterial and mixed venous oxygen saturation • RHC can assess vaso-reactive response • shown in only 1015% of patients • sustained response is shown in less than 7% of patients RIGHT HEART CATHETERISATION-THE GOLD STANDARD VASOREACTIVITY • Testing is done with right heart catheterization • Defined as mean pulmonary artery pressure decrease by at least 10 mm Hg to a level below 40 mm Hg, with no decrease in cardiac output. • Agents approved - Epoprostenol IV - Adenosine IV - Nitric oxide inhaled FUNCTIONAL ASSESSMENT • WHO functional classification • 6 minute walk test (6 MWT) - Reliable, validated - Correlates with survival - Common endpoint in therapeutic trials WHO FUNCTIONAL CLASSIFICATION • There are four WHO functional classes (WHO FC) • WHO FC I being the least severe • WHO FC IV being the most advanced Different classes reflect the impact on a patient’s life in terms of physical activity and symptoms WHO FUNCTIONAL CLASSIFICATION • • • • Class I Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause dyspnea or fatigue, chest pain or near syncope Class II Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope Class III Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnea or fatigue, chest pain or near syncope Class IV Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may even be present at rest 6 MINUTE WALK TEST • The 6-minute walk test (6MWT) is an evaluation of exercise capacity in patients with PAH and is used to determine the functional class and is also a prognostic indicator • The 6-MWT is a critical endpoint in clinical studies assessing therapeutic options • The 6-MWT should be performed under supervision and according to a standardised protocol BIOMARKERS • N- TERMINAL Pro BNP • Hyponatremia - Neuro hormonally released Vasopressin induced - Strongly associated with RHF and poor prognosis TREATMENT • There is currently no cure for PAH • Prognosis is influenced by the status of WHO FC when treatment is started – those who start therapy in WHO FC I or II demonstrate a better prognosis than those whose therapy is started in the more severe stages1 • By recognizing and treating patients as early as possible, disease progression may be delayed • Without treatment, patients in WHO FC II can rapidly deteriorate within 6 months to more advanced PAH as evidenced by progression of symptoms. TREATMENT STRATEGY • • • • Prevention Screening of high risk patients Optimize therapy for related disease Supportive treatment ( aimed at consequences of PAH) • Vascular targeted therapy (aimed at reducing/ reversing vasoconstriction, proliferation) • Surgical ( atrial septostomy, lung transplantation) SUPPORTIVE Anticoagulants - warfarin for prevention against in situ thrombosis INR goal is 2-2.5 Diuretics – for treatment of right heart failure Oxygen therapy – to maintain oxygen saturation at >90% at all times Pneumococcal and yearly influenza vaccination Digoxin Avoidance of High altitudes Air travel Pregnancy NSAID Tobacco use Heavy exertion Beta blockers Decongestant medications High Sodium diet Appetite suppressants VASCULAR TARGETED THERAPIES • Prostacyclin pathway Beraprost (PO) Ilioprost (Inhaled) Epoprostenol (IV) Treprostinil (SQ,IV, inhaled) • NO pathway Dipyridamole(PO) Nitric oxide (inhaled) Sildenafal (PO) Taladafil (PO) • Endothelin pathway Ambrisentan (PO) ER-A antagonist Bosentan(PO) non selective CALCIUM CHANNEL BLOCKERS • Never given empirically • Reserved for “responders” -Near normalization (PAP to 40 mm) and normal CO -High dose nifedipine or diltiazem -Only half of responders will have long term benefit -Response monitored and with deterioration treatment escalated Survival in patients with IPAH based on acute vasoreactivity and treatment with warfarin. In patients who were not vasoreactive, warfarin was associated with a modest survival advantage versus nonreactive patients who were not on warfarin. (Redrawn from Rich S, Kaufmann RN, Levy PS: The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J COMBINATION THERAPY • Maximize therapy while minimize toxicity • 8 trials with 1600 patients published various combinations 6MWT as the primary endpoint 12 -16 weeks showed only modest improvement • Recommended for Class III and IV who fail to improve or class II with deterioration SURGICAL OPTIONS • Atrial septostomy -Reduced RVEDP, increased CI, increased exercise capacity at expense of decreased PaO2 -Option in patients unresponsive to max Rx -Bridge to transplantation • Lung transplantation -Typically bilateral lung transplantation -Heart-lung transplantation in patients with PAH secondary to congenital heart disease • Pulmonary thromboendarterectomy for patients with CTEPH TREATMENT ALGORITHM FOR PAH PROGNOSIS • 15 % one year mortality on modern therapy • Markers of poor outcome -High functional class -Poor performance on 6 MWT -High RA pressure -Significant RV dysfunction -RV failure -Low Cardiac index -Elevated N terminal Pro-BNP -Hyponatremia PAH and PREGNANCY • Mortality rate approaches 30% • Contraception is strongly recommended in women of child bearing age with PAH • Mechanical contraception (IUD) or surgical sterilization is recommended Summary • PAH is a complex disease involving vascular proliferation, inflammation & remodeling • Unexplained dyspnea should be worked up in a stepwise fashion • Good history and physical can give clues towards the diagnosis and etiology • Diagnosis in early stage leads to early treatment and better outcome. • Echocardiogram is an internationally recommended screening tool. • Right heart catheterization is needed for definitive diagnosis • Calcium channel blockers are to not be used empirically • Vascular targeted therapies now available and improve quality of life and mortality. • Mortality remains high even with the modern pharmacologic interventions • Pregnancy is contraindicated because of high mortality REFRENCES • Pulmonary Board review 25th edition • www.phaonline.org • Murray and nadel’s “Textbook of respiratory medicine” 5th edition. • Gilead sciences inc.