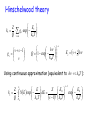Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Wave–particle duality wikipedia , lookup
X-ray photoelectron spectroscopy wikipedia , lookup
Renormalization wikipedia , lookup
Molecular Hamiltonian wikipedia , lookup
Scalar field theory wikipedia , lookup
Theoretical and experimental justification for the Schrödinger equation wikipedia , lookup
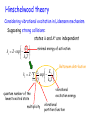
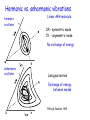
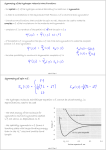
This course material is supported by the Higher Education Restructuring Fund allocated to ELTE by the Hungarian Government Theories of unimolecular reactions Ernő Keszei Eötvös Loránd University, Institute of Chemistry Overview – Introduction – Historical overview – Lindemann theory – Some aspects of statistical physics – RRK theory – RRKM/QET theory – TST and activation entropy – IVR: internal vibrational relaxation – (Some) experimental evidence of TST – Recent developments in RRKM theory Introduction Unimolecular reaction – formal description A products d [A] k [A] dt Typical unimolecular reactions – photochemical (laser induced) – isomerisations – gas phase reactions – very low pressure reactions (mass spectrometer, upper atmosphere) Historical overview – early 20th century: Gas phase unimolecular reactions (mostly pyrolysis) Many 1st order reactions were thought to be unimolecular Problem: how molecules acquire activation energy? – Perrin’s suggestion (1919) molecules excited by radiation – opposed by many chemists – the rate should depend on surface/volume ratio – it should not depend on the pressure – there should be a difference between dark / sunlight conditions Perrin’s idea has a revival recently: IR laser activation Fourier-transform mass spectrometry Historical overview ”inexplicable” experimental evidence Decomposition of propionaldehyde and ethers (Hinshelwood) Decomposition of azo-methane 1927: experiments of Ramsperger high pressure: first order kinetics low pressure: second order kinetics The rate increases with increasing temperature, but first order kinetics is not in accordance with collisonal activation Pilling & Seakins 1995 Lindemann theory (1922) A+M A*+ M A* 𝑘1 𝑘−1 𝑘2 A* + M activation (via collisions) A +M deactivation (via collisions) P (products) decomposition (spontaneous) M: any molecule (A, P or inert gas molecules added) Rate equation for the energised molecules: d [A*] k1[A][M ] k 1[A*][M ] k 2 [A*] dt formation removal Considering A* as a steady-state component: k1[A][M ] k 1[A][M ] k 2 [A*] k1[A][M ] [A*] k 1[M ] k 2 Lindemann theory A+M A*+ M A* 𝑘1 𝑘−1 𝑘2 A* + M activation A +M deactivation P (products) decomposition k k [M ][A] k uni [A] 2 1 k 1[M ] k 2 k uni k 2 k1[M ] k 1[M ] k 2 Low pressure limit: k 1 M k 2 M k2 k 1 High pressure limit: k 1 M k 2 M k2 k 1 k uni k1 M k uni k 2 k1 k 1 2nd order 1st order In accordance with experiments Lindemann theory A+M A*+ M A* 𝑘1 𝑘−1 𝑘2 A* + M activation A +M deactivation P (products) decomposition Low pressure: k uni k1 M 2nd order k1M k2 : accumulation of energised molecules Activation is the rate determining step High pressure: k uni k 2 k1 k 1 1st order k1M k2 : fast removal of energised molecules via collisions Product formation is the rate determining step, but 𝑘uni ≠ 𝑘2 lg (kuni / s–1) Comparison with experiment lg k k k 2 k1 k 1 kuni extrapolated to infinite pressure k k 2 k1 kk 2 1 2 k 1 k 2 M 1 2 2k 1 lg (k / 2) [M]1/ 2 k 2 k k 1 k1 [M]1/2 : where kuni = k / 2 lg ([M]1/2) from experiment: k and [M]1/2 from collision theory: 𝑘1 = 𝑍𝑒 lg ([M] / mol [M]1/ 2 k k1 dm–3 Calculated [M]1/2 = 𝐸 − 𝑅𝑇 𝑘∞ 𝑘1 We can compare [M]1/2 and [M]1/2 Comparison with experiment from experiment Reaction calculated T/K Z / s–1 E / kJmol–1 [M]1/2 [M]1/2 760 3·1015 276 3·10–4 2·104 720 4·1015 267 1·10–5 2·104 670 3·1015 256 1·10–6 1·104 MeNC → MeCN 500 4·1013 161 4·10–3 2·102 EtNC → EtCN 500 6·1013 160 4·10–5 4·102 N 2 O → N2 + O 890 8·1011 256 8·10–1 4 2 + Pilling & Seakins 1995 Many orders of magnitude differences! Comparison with experiment k k 2 k1 M k 1 M k 2 1 k 1 M k 2 k k 2 k1 M 1 k 1 1 1 1 k k1k 2 k1 M k k1 M 1 1 1 1 k k k1 M 1 1 𝑣𝑠. 𝑘 M should be linear — it is not Pilling & Seakins 1995 Lindemann theory — summary • Two-step procedure with collisional activation • Steady-state approximation for the energised species Advantage: • A simple and qualitatively fairly correct picture Disadvantages: • underestimation of k1: neglecting molecular degrees of freedom storing energy • neglecting internal vibrational relaxation in product formation Improvements to Lindemann theory Avoiding underestimation of k1 — vibrational excitation processes are more effective supposing strong collisions (independent states before and after) equivalent harmonic oscillators for vibrations (Hinshelwood, 1927) anharmonic oscillators, quantum mechanical description (Marcus, 1952) — stepwise energy uptake replaces strong collisions ”master equations” (Tardy, Rabinovitch, Schlag, Hoare, 1966) Improvements to Lindemann theory Considering the role of IVR in decomposition — equivalent oscillators: RRK theory (Rice, Ramsperger, Kassel, 1928) semi-classical treatment of oscillators, plus role of rotation: RRKM theory (Marcus, 1951) — more precise calculation of density of states & partition functions for vibrational states (Whitten-Rabinovich, 1959) direct counting of vibrational states (Current, Rabinovich 1963; Beyer, Swinehart, 1973) Hinschelwood theory Considering vibrational excitation in Lidemann mechanism Supposing strong collisions: states A and A* are independent E k1 Z exp k BT minimal energy of activation Ev gv k1 Z exp v m Q k BT quantum number of the lowest excited state multiplicity Boltzmann distribution vibrational excitation energy vibrational partition function Density of states Discrete states: the number of quantum states at the same energy (also called degeneration) e. g. Two oscillators with the same frequency: Ev=(v+1/2)h Ev 3,0 2,1 1,2 2,0 1,1 0,2 1,0 0,1 0,3 0,0 4 3 2 1 v1,v2 : vibrational quantum numbers Degeneration of s oscillators with the same frequency: (v s 1)! gv v!( s 1)! Details of counting states Let us distribute v quanta of vibration among s oscillators: → equivalent to the distribution of v pebbles among s boxes equivalent to the distribution of v pebbles and s – 1 bars Degeneration of s oscillators with the same frequency: (number of permutations with repetition) v s 1 v s 1! g v v v !s 1! Density of states Continuous approximation Continuous states: the density of continuous states at the same energy notation: ρ(E ) Equivalent to the number of states between energies E and E + dE E' Definitions: number of states between 0 and E’ : N ( E ' ) ( E )dE 0 density of states at energy E : (E) dN ( E ) dE Density of states at energy E 1D translation 2ma 2 (E) Eh 2 a : 1D box length 3D translation 4 32 (E) 3 m 2E V h V : volume rotation (linear molecule) (E) 1 B B : rotational constant E Ba Bb Bc rotation (3D molecule) (E) 2 general expression: ( E ) E ( s / 21) Ba , Bc , Bd : rotational constants s : number of degrees of freedom Vibrational density of states at energy E Classical harmonic oscillators: E E s 1 s ( s 1)! h i N E Es s s! h i underestimation by several orders of magnitudes i 1 i 1 z Semiclassical harmonic oscillators (no quantum effects but zero-point energy): E ( E E z ) s 1 s ( s 1)! h i i 1 N E ( E Ez ) s s s! h i i 1 Ez h i 1 i 2 more moderate underestimation Hinschelwood theory Ev Z k1 g v exp Q vm k BT v s 1 g v v h Q 1 exp k T B s Ev v 12 h Using continuous approximation (equivalent to h k BT ): E Z Z E0 dE k1 N E exp Q E0 s 1! k BT k BT s 1 E0 exp k BT Hinschelwood theory E k L Z exp k BT Z E0 k1 s 1! kBT k1 1 E0 k L s 1! k BT As E0 k BT s 1 E0 exp k BT s 1 1 E0 1 k BT Even so, an unrealistically large s should be given for large molecules Vibrational density of states at energy E Anharmonic quantum oscillators: no closed (analytical) expression for the number of states Solution: direct counting of states (I)=[1,0,0,0….0] FOR J=1 TO s FOR I=(J) TO M (I)=(I)+(I - (J)) NEXT I NEXT J - counting array initiation s: number of oscillators M = maximum of energy (J) - oscillator frequency, cm–1 loop for energy loop for the oscillators No serious underestimation — the problem concerning k1 is more or less solved RRK theory (Rice, Ramsperger and Kassel; 1928) For the energised molecule to dissociate, excess energy should be concentrated in the critical vibrational modes. New mechanism: A+M A*+ M A* A‡ 𝑘1 𝑘−1 𝑘∗ 𝑘‡ A* + M activation (via collisions) A +M deactivation (via collisions) A‡ formation of the transition state (IVR) P (products) decomposition (spontaneous) IVR is much slower than the decomposition: k* << k‡ Considering A‡ as a SS component: 𝑘 ∗ A∗ = 𝑘 ‡ A‡ ‡ A 𝑘∗ = 𝑘‡ ∗ A RRK theory Underlying conditions: oscillators of the same frequency equal probability of all (vibrational) states The transition state is formed, if there is enough energy concentrated on the critical vibrational mode(s) which results in the product formation The critical number m of vibrational quanta should be accumulated in the critical vibrational mode Pilling & Seakins 1995 RRK theory 𝑘∗ = A‡ ‡ 𝑘 ∗ A = 𝑘 ‡ 𝑃𝑚𝑣 𝑃𝑚𝑣 is the probability that m quanta out of v are on the critical mode v s 1 v s 1! Recall: g v s 1!v ! v v–m m g vm v m s 1 v m s 1! v m s 1!v m ! v, m and v–m >> s g vm v! v m s 1 ! v m P v s 1! v m ! gv v s 1 s 1 v m m 1 v s 1 RRK theory Rewriting 𝑘 = 𝑘 ∗ ‡ ∗ ‡ 𝑃𝐸𝐸0 = A‡ 𝑘 =𝑘 Recall: 𝑃𝑚𝑣 = A∗ 𝑘 1− ‡ 1− 𝑚 𝑠−1 𝑣 𝐸0 𝑠−1 𝐸 for continuous energy: 𝑃𝐸𝐸0 = and 𝑘 ∗ = 𝑘 ‡ 1− 𝐸0 𝑠−1 𝐸 A‡ A∗ 𝑘 ∗ = 𝑘 ‡ 𝑃𝐸𝐸0 Resulting expression for k2 (product formation): 𝑘2 𝐸 = 𝑘 ‡ 𝐸0 1− 𝐸 𝑠−1 The reaction rate is energy dependent; it increases with increasing energy RRK theory 𝑘2 𝐸 𝑘‡ Limit case (Lindemann theory) 𝑬 𝑬𝟎 Pilling & Seakins 1995 RRK theory — summary of results 𝐸 𝑘B 𝑇 𝑘2 𝐸 = 𝑘 ∗ (𝐸) = 𝑘 ‡ k uni E0 𝐸0 1− 𝐸 𝑠−1 k1 ( E ) k 2 ( E )M dE k 1 M k 2 ( E ) Energy / cm–1 𝑍𝑁 𝐸 − 𝑘1 𝐸 = 𝑒 𝑄 compared to the Lindemann expression: k uni k 2 k1 M k 1 M k 2 Reaction rate (arbitrary units) Pilling & Seakins 1995 RRK theory — summary of concepts • Two-step procedure with collisional activation • Steady-state approximation for both the energised species and the transition state • k1 estimated by distributing energy over vibrational modes (presupposing strong collisions) • k2 estimated by the probability of TS formation with IVR (fast flow of energy between vibrational modes) Approximations used: oscillators of the same frequency continuous energy distribution RRKM theory (Marcus; 1952) k1 calculated using quantum statistical method Proper (different!) frequencies of A* and A‡ used Rotational modes of TS are properly considered Introduction of constant and variable energy: constant energy: zero point energy and translational energy variable energy participates only in IVR New mechanism: A+M A*(E ) + M A*(E ) A‡ 𝑘1 𝑘−1 𝑘∗(𝐸 ′ ) 𝑘‡ E: total energy A*(E ) + M activation (via collisions) A +M deactivation (via collisions) A‡ formation of the transition state (IVR) P (products) decomposition (spontaneous) E’: variable energy RRKM theory — energy landscape total energy 𝐸𝑟‡ Er E0 ”activation energy” stable molecule 𝐸𝑡‡ E‡ E Ev 𝐸𝑣‡ zero point energy of transition state E0 zero point energy of stable A E Er Ev total energy rotational energy energy of vibrations and internal rotations transition state E‡ 𝐸𝑟‡ 𝐸𝑣‡ max. available energy 𝐸𝑡‡ rel. translational energy rotational energy energy of vibrations and internal rotations RRKM theory — role of rotation Conservation laws: both energy and momentum are conserved 𝐸𝑟‡ Er E‡ Angular momentum: L = Iω E 1 Rotational energy: 𝐸𝑟 = 𝐼𝜔2 2 I : moment of inertia; ω: angular velocity 𝐸𝑡‡ 𝐸𝑣‡ Ev E0 Formation of TS: I increases → Er decreases I << I ‡ → 𝐸𝑟 − 𝐸𝑟‡ = 𝐸𝑟 1 − 𝐼 𝐼𝑟‡ Excess 𝐸𝑟 − 𝐸𝑟‡ becomes vibrational energy Ev Loosening of internal bonds result in increase of vibrational energy RRKM / QET theory E = Ev + Er Iternal degrees of freedom: - active : - adiabatic: free energy flow (vibration and internal rotation) rotational quantum state cannot change (rotation of the whole molecule) Expression of the rate constant: # # # # # E E E t # v k Ev ,Et Ev Ev Number of states in the transition state Number of states for the active degrees of freedom Integrating for all energies, we get the rate constant: k EV ‡ # E t d t EV N # E# EV Relation of RRKM to RRK theory Number of states and density of states for classical harmonic oscillators: E E s 1 ( s 1)!(h ) s N E Es s!(h ) s (The n. d. f. is one less for the TS than for the reactant molecule.) Substitute them into the RRKM equation: k E N # ( E E0 ) E ( E E0 ) s 1 1 ( s 1)!(hv) s 1 E0 1 s 1 (E ) h E ( s 1)!(hv) s s 1 We get (back) the RRK equation The role of activation entropy (S#) q# U U S kb ln q T # # S kbT A exp R h # A – preexponential q – partition function U – zero point energy T – temperature S# > 0 fuzzy transition state e.g. bond braking S# < 0 “closed” transition state e.g. rearrangement 12 Example: 10 8 - Butyl benzene radical decomposition: MS detection: internal energy can be estimated from fragment ion ratio lg(k) formation of propene and propyl radical 6 4 2 0 -2 -4 -6 2 4 6 8 Internal energy(eV) (eV) Belsõ energia Keszei 2015 10 Relation of microcanonical to canonical rate constant k(E) – microcanonical k(T) – canonical rate constant High pressure: equilibrium distribution is maintained canonical energy distribution: Pth ( E ) ( E )e ( E )e E k bT E k bT ( E )e 0 k (T ) Pth ( E )k ( E )dE 0 k (T ) E0 ( E )e Q E k BT density of states Boltzmann konstant internal energy temperature (E) kB E T N ( E E0 ) 1 dE N # ( E E0 )e E Q E0 # E k BT dE Q E k bT Presuppositions of RRKM/QET theory Random distribution of states Necessary condition is that the IVR be much faster compared to the decomposition rate All TS configurations transform into products A good approximation for relatively large molecules, and relatively low energies above the activation threshold. Reaction coordinate is orthogonal to other coordinates: it is separable from them It is fulfilled at low energies where vibrational modes can be described by non-coupled normal modes. With increasing energy, the probability of coupling increases. Harmonic vs. anharmonic vibrations harmonic oscillator Linear ABA molecule QR - symmetric mode TS - asymmetric mode No exchange of energy anharmonic oscillator Lissajous motion Exchange of energy between modes Pilling & Seakins 1995 Role of IVR (Intramolecular Vibrational Relaxation or Redistribution - IVR) Excitation: typically one single vibrational mode This localised energy should be redistributed into other (critical) vibrational mode(s) If IVR is fast compared to the reaction, random energy redistribution can be applied If the reaction is fast compared to IVR, random energy redistribution cannot be applied. State dependent rate constants should be calculated, and all relevant states should be considered. Experimental testing of IVR Rabinovitch et al., JPC 78, 2535, 1974 Ratio of the two products: CF2 CF + CH2 With increasing pressure the ratio increases CF2 → more frequent collisions more effective deactivations not enough time for IVR CF CF CD2 CF2 CH2 1 CF2 CF CD2 IVR rate is ~ 1 ps CF2 CD2 1:1 at low pressures: perfect IVR Model calculations: CF 2 CF + CF2 CH2 dissociation at the addition site CD2 CF CF + CF2 CF2 CH2 at the other site A flaw in RRKM theory Calculations are made using harmonic oscillators In principle, no exchange of vibrational energy is possible But for RRKM theory to work, fast IVR is needed However, RRKM theory performs quite well! Possible explanation: Small deviations from harmonicity of the anharmonic oscillators does not affect the precision of the results much but is just enough to maintain fast energy transfer between vibrational modes Indirect evidence of the transition state Moore et al., Science 256, 1541 (1992) Dye laser excitation and detection 1. pulse → S1 fast ISC → T1 Energy / 103 cm–1 CH2CO CH2 + CO (above activation energy) 2. pulse: detection of CO Reaction coordinate Pilling & Seakins 1995 k (E) is expected to increase stepwise with increasing energy Indirect evidence of the transition state Measured intensity / arbitrary units (a) disappearance of ketene calculated (b) production of CO measured (c) production of CO calculated (d) disappearance of ketene measured Good correlation between experiment and model calculations Energy / cm–1 Pilling & Seakins 1995 Further improvements in RRKM theory — correct account of anharmonicity (especially for reactions with several minima in the PES) — correct account of vibrational-rotational couplings (especially important for smaller molecules) — Variational Transition State Theory (VTST) If no saddle point is found on the Potential Energy Surface, reaction rate should be calculated at the minimum of N‡(E‡) — Orbiting Phase Space Theory For reactions, where the opposite reaction (recombination or association) does not have an activation energy. (There is no need to take into account a transition state.) — Using master equations (if the approximation of strong collisions does not apply) Using the master equation If collisions are not strong, we should consider all subsequent collisions Unlike Lindemann theory, where only ground state and energised state are considered, we should follow all vibrationally excited states Formal mechanism used: A(i) + M A( j) + M A( j) 𝑍𝑃𝑗,𝑖 𝑍𝑃𝑖,𝑗 𝑘𝑗 A( j) + M activation A(i) + M deactivation P (products) decomposition 𝑃𝑗,𝑖 is the probability of transition from state i to state j 𝑃𝑖 is the probability of finding the molecule in state i Z is the collision number Using the master equation Equations to describe probabilities of transitions population at level j (probability: Pj) n Z Pi , j j 0 k (i) decomposed population n Z Pj ,i j 0 population at level i (probability: Pi) Transfer equation: Both i and j can have any value between 0 and n n n dPi (t ) Z Pi , j Z Pj ,i k (i) dt j 0 j 0 Master equation n The probability of being in any of the states: P j 0 j ,i 1 n dPi (t ) Z Pi , j 1 k (i ) dt j 0 a homogeneous linear system of ordinary differential equations: one equation for each state i It can be solved with any standard solution method: (finding eigenvalues and eigenvectors is the usual one) Further reading – M. J. Pilling, P. W. Seakins: Reakciókinetika, Nemzeti tankönyvkiadó, Budapest, 1997 – T. Baer , W. L. Hase: Unimolecular Reaction Dynamics, Oxford University Press, 1996 – J. I. Steinfeld, J. S. Francisco, W. L. Hase: Chemical Kinetics and Dynamics, Prentice Hall, 1989 – P. J. Robinson, K. A. Holbrook: Unimolecular Reactions, Wiley, 1972 Acknowledgements Figures and tables marked as Pilling & Seakins 1995 are reproduced from M. J. Pilling, P. W. Seakins: Reaction Kinetics, Oxford University Press, 1995 END of the lecture Theory of unimolecular reactions Thank you for your attention ! This course material is supported by the Higher Education Restructuring Fund allocated to ELTE by the Hungarian Government