Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
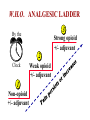
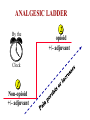
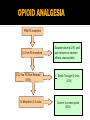
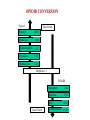
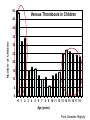
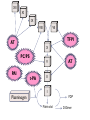
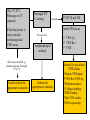
Pain Management (2 phase approach) Neonatal Screening for Hemoglobinopathy Epistaxis: The Hematological Angle Thrombophilia Investigation: When and How Hadi Sawaf MD A new BOXED WARNING, FDA’s strongest warning, will be added to the drug label Health care professionals should prescribe an alternate analgesic for post-operative pain control in children who are undergoing tonsillectomy and/or adenoidectomy. Codeine should not be used for pain in children following these procedures Codeine • Codeine effect is dependent upon its conversion to morphine by the hepatic cytochrome P450 2D6 (CYP2D6). • Some people have DNA variations that make this enzyme more active, causing codeine to be converted to morphine faster and more completely than in other people • High levels of morphine can result in breathing difficulty, which may be fatal. • Taking codeine after tonsillectomy and/or adenoidectomy may increase the risk for breathing problems and death in children who are “ultra-rapid metabolizers.” • For other types of pain in children, codeine should be used if the benefits are anticipated to outweigh the risk Codeine Eliminate the use of codeine in pediatric patients <18 years of age in inpatient and outpatient settings throughout Ascension Health Will undertake a codeine phase-out over an approximate 90-day period W.H.O. ANALGESIC LADDER 3 By the Strong opioid +/- adjuvant 2 Clock 1 Non-opioid +/- adjuvant Weak opioid +/- adjuvant ANALGESIC LADDER By the Clock 1 Non-opioid +/- adjuvant 2 opioid +/- adjuvant Oral Dose Medicine Neonate: 0 to 29 days Infants: 30 d to 3 mos Infant 3-12 mos or child 1-12 yrs Maximum Daily dose Acetaminophen 5–10 mg/kg every 6–8 hr 10 mg/kg every 4–6 hr 10–15 mg/kg every 4–6 hrs 4 doses/day 5–10 mg/kg every 6–8 hrs Child: 40 mg/kg/day Ibuprofen Intravenous Acetaminophen • Approved by FDA for the use in children 2 years of age and older • Maximum serum concentration after IV acetaminophen was 70% higher than the same dose given orally • Does not appear to increase the risk for hepatotoxicity • Dose: 12.5 mg/kg IV every 4 hours or 15 mg/kg IV every 6 hours, with a maximum dose of 75 mg/kg every 24 hours. Opioid Brand Names Generic Name fentanyl hydrocodone hydromorphone Brand Name Duragesic Norco, Vicodin Dilaudid, Exalgo morphine oxycodone Astramorph, Avinza OxyContin, Percocet Medicine Route of administration Starting dose Morphine Oral (immediate release) 1–2 years: 200–400 mcg/kg Q 4 hrs 2–12 years: 200–500 mcg/kg Q 4 hrs (max 5 mg) IV injection 1–2 years: 100 mcg/kg Q 4 hrs 2–12 years: 100–200 mcg/kg Q 4 hrs Oral (immediate release) 30–80 mcg/kg Q 3–4 hrs (max 2 mg/dose) Oral (prolonged release) 15 mcg/kg Q 3–6 hrs Hydromorphone *opioid-naive children OPIOID ANALGESIA PRN PO morphine Q 4 hrs PO morphine Q 12 hrs PO Slow Release (50%) IV Morphine (1:3 ratio) Escalate dose by 25% until pain relieved or adverse effects unacceptable + Break Through Q 4 hrs (10%) Convert to a new opioid (50%) OPIOID CONVERSION Opioid Most Potent Fentanyl 100:1 Methadone 10:1 Hydromorphone 5:1 Oxycodone 1.3:1 Hydrocodone 1.2:1 Morphine 1:1 NSAIDS Ibuprophen 1:40 Naproxen 1:50 ASA Least Potent 1:130 Acetamenophen 1:130 Amino Acid Disorders: X 14 - April 2014-CCHD - October-2011 SCIDCCHD - October 2007 CF and Hearing Fatty Acid Oxidation Disorders: X 12 Organic Acid Disorders: X13 Hemoglobinopathies: • S/Beta thalassemia • S/C disease • Sickle cell anemia • Variant hemoglobinopathies • Hemoglobin H disease - October 2004-HCY, CIT, ASA - April 2005-31 MS/MS Disorders - April 2003-MCAD - July 1993-CAH - October 1987-Niotinidase Deficiency, - MSUD and Hemoglobinopathy - Spring 1985-Galactosemia - June 1977-CH Endocrine Disorders: X2 Other Disorders: X7 - August 1865-Phenyketonia In 2011: 1 in 364 screened African American newborns and 1 in 1,907 screened newborns were diagnosed with SCD. • An additional 2,817 newborns were identified as having sickle cell trait based on initial screening results. • 61 newborns were diagnosed with SCD Incidence: In 2011: 1 in 364 screened African American newborns and 1 in 1,907 screened newborns were diagnosed with SCD. 61 newborns were diagnosed with SCD SCD Subtype, Confirmed Cases, 2011 Subtype N % Hemoglobin SS (HbSS) 33 54 Hemoglobin SC (HbSC) 20 33 Sickle Beta Thal Plus 8 13 The purpose of newborn hemoglobinopathy screening is to detect sickle cell disease (HGB SS, SC, Sβ° and Sβ⁺) Most common, abnormal (non sickle) HGBs: C, D, E and Bart’s hemoglobin Methodology : high performance liquid chromatography (HPLC) and isoelectric focusing. A confirmatory hemoglobin electrophoresis is required before age 3 months Neonatal Screening for Hemoglobinopathy Other hemoglobins are reported as “V”. They invariably have no or minimal clinical or genetic significance and are not report to parents Hemoglobins are generally reported in decreasing order of concentration (F>A>S) Newborn hemoglobinopathy screening will not identify beta thalassemia trait DIAGNOSIS CONFIRMATORY FAMILY STUDIES Sickle Cell Anemia (SS) FS Both parents AS Sickle Beta Thalassemia Zero (Sβ°) FS One parent AS One parent AA with elevated HB A2 Sickle Cell with HPFH F FS One Parent AS One parent AF with Hb F approx. 20‐30% Education Session Completion Sep 2011- Oct 2012 • • • • Session 1 SCD overview Early health problems Sickle cell trait vs disease Session 2 • Transmission of SCD • Types of SCD • Late health problems 50% 40% 30% 20% 10% 0 2 sessions completed 1 session completed Previously educated Refuse or unable Unknown Sickle Cell Disease in Michigan Percent of Children with SCD with TCD Screen by Year and Gender Percent of Children with SCD Receiving Antibiotic Prophylaxis 100% 25% 80% 20% 60% 15% Within 120 days 40% Female 10% Before 5 mos Male 20% 5% 0 0 2007 2008 2009 Birth Year 2010 2011 2008 2009 Birth Year 2010 2011 Sickle Cell Disease Confirm diagnosis Penicillin prophylaxis Disease Education Referral to Pediatric Hematology/Oncology RESULT DIAGNOSIS ACTION REQUIRED FAS Sickle cell trait Clinically benign but genetically significant No confirmatory testing Genetic counseling FAC Hemoglobin C trait Clinically benign but genetically significant No confirmatory testing Genetic counseling FAD Hemoglobin D trait Clinically benign but genetically significant No confirmatory testing Genetic counseling FAE Hemoglobin E trait Clinically benign but genetically significant No confirmatory testing Genetic counseling RESULT DIAGNOSIS ACTION REQUIRED FAV Fetal hemoglobin, normal adult and an unidentified hemoglobin variant Most likely clinically Physician of record insignificant hemoglobin responsible for reassuring variant the parent that this is clinically insignificant. No confirmatory testing required. FA-Bart’s Fetal Hemoglobin, Hemoglobin A and Bart’s Hemoglobin Hemoglobin H disease Alpha Thalassemia Trait MDCH will send specimen to reference lab for further analysis. SCDAA will provide information to parents and physician of record for clinically significant findings Thrombophilia Testing Who and How Definition: Thrombophilia an inherited or acquired abnormality of hemostasis predisposing to thrombosis Some form of Thrombophilia can be identified in approximately half of patients presenting with VTE Thrombophilia factors may enhance the risk of recurrent thrombosis. Therapeutic and prophylactic measures are not necessarily different for children with or without thrombophilic risk factors Individualized approach is warranted Incidence: 5 cases per 10,000 children per year Mortality rate for major vessel thrombosis is 1% - 4% Thrombus recurrence: 6.5% to 21% of children with VTE Many cases of DVT and pulmonary embolism (PE) go unrecognized in part due to a low index of suspicion for young patients 50 Venous Thrombosis in Children 45 40 Number of Children 35 30 25 20 15 10 5 0 <1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Age (years) From Canadian Registry Venous Thromboembolism in Children Site Extremity Superior vena cava Splenic vein Hepatic vein Renal vein Portal vein Pulmonary artery Cerebral sinus Presentation Dx D-Dimer Pain Swelling Discoloration Tissue damage Doppler ultrasound MR venography (MRV) CT angiography (CTA) Venography Echocardiography XII XI IX VIII VII TFPI AT X PC/PS PAI V t-PA AT II I Plasminogen FDP Fibrin clot D-Dimer Risk factors for venous thrombosis Procoagulant Anticoagulant Other High levels of factor VIII Antithrombin deficiency Factor V Leiden (FVL) High levels of factor IX Protein C deficiency Antiphospholipid syndrome High levels of factor XI Protein S deficiency Hyperhomocysteinemia High levels of fibrinogen Prothrombin 20210A Dysfibrinogenemia Prevalence of Major Hypercoagulable States Hypercoagulable State General Inheritance Risk for 1st Patient Population VTE % with % single VTE % Thrombophilic Families Factor V Leiden 3-7 AD 3-5 20 50 Prothrombin G20210A 1-3 AD 2-3 6 18 Anti-thrombin deficiency 0.02 AD 5-10 1 4-8 Protein C deficiency 0.2-0.4 AD 4-6.5 3 6-8 Protein S deficiency 0.02-0.04 AD 1-10 1-2 3-13 Homocysteinemia 5-10 AR 2-3 10-25 N/A Antiphospholipid antibodies 0-7 Acquired 1-8 5-15 N/A Some numbers for U.S. population; approximation) A. Hemophilia 28,000 have hemophilia A 5,000 have hemophilia B B. Thrombophilia 13 million are heterozygous for factor V Leiden 170,000 are homozygous for factor V Leiden 5,6 million are heterozygous for prothrombin 20210 mutation 28,000 are homozygous for prothrombin 20210 mutation 70,000 are heterozygous for factor V Leiden plus heterozygous for prothrombin 20210 mutation Number of Children Who should be tested? VTE at a young age with Spontaneous Recurrent Unusual site Positive family history Testing is probably not helpful Catheter related thrombosis Before initiation of contraceptive Asypmtomatic child with positive family history Incidence: 4-21.3% Multitrait thrombophilia Spontaneous VTE Homozygous factor V Leiden Prothrombin gene mutation Antiphospholipid Abs Elevated D-dimer at the end of anticoagulant therapy *The recurrence risk did not decrease with increased duration of anticoagulation Thrombophilia Diagnostic Laboratory Studies Level (I )Testing Level (II) Testing Level (III)Testing Thrombophilia Laboratory Tests Factor V Leiden PCR or clotting assay (APC res.) Prothrombine G20210A PCR Antithrombin deficiency Chromogenic or clotting assay Protein C deficiency Chromogenic or clotting assay Protein S deficiency Clotting assay or immunologic Homocysteinemia Fasting homocysteine Antiphospholipid antibodies Clotting or chromogenic assay Elevated factor VIII Clotting assay Dysfibrinogenemia Clotting or immunologic assay Elevated factor IX, XI Clotting assay Case History: A 5- year-old boy presents to clinic with his parents who are concerned about his 3-month history or recurrent nosebleeds On 2 occasions he was send home from school because of the nose bleed His physical examination was entirely normal with no petechia or bruises noted. When in your office he developed epistaxis that lasts about 15 minutes. His platelet count is 178,00,000. His complete blood count and smear review were normal ▸ Inflammation – URI – Allergic rhinitis – Foreign body – Vasculitis ▸ Trauma – Nose picking – External trauma ▸ Anatomic – Septal deviation ▸ Medications – Topical steroid spray – Nasal decongestants – Anticoagulants ▸ Hematologic – Idiopathic thrombocytopenic purpura – Von Willebrand disease – Hemophilia ▸ Neoplasms – Benign ∘ Nasopharyngeal angiofibroma ∘ Pyogenic granuloma ∘ Inverted papilloma – Malignant ∘ Rhabdomyosarcoma ∘ Lymphoma ▸ Vascular abnormalities – Hereditary hemorrhagic telangiectasia – Hemangioma ▸ Idiopathic Epistaxis unrelieved by 10 minutes Epistaxis requiring ER visit, or blood transfusion History other bleeding manifestations Bleeding from trivial wounds >15 mints Bleeding from dental procedures > 1 day or requiring a blood transfusion Heavy, prolonged or recurrent bleed after surgical procedure Heavy menses Family history of a bleeding disorder Epistaxis Scoring system Component Score Frequency 5-15/yr 16-25/yr >25/yr 0 1 2 Duration < 5 min 5-10 min >10 min 0 1 2 Amount < 15 ml 15-30 ml >30 ml 0 1 2 Epistaxis history/age < 33% 33-67% >67% 0 1 2 Site Unilateral Bilateral 0 2 Mild: 0-6 Severe: 7-10 Likelihood Ratio for VWD 10000 1000 100 0.10 1 0.1 0.01 0.001 -3 -2 -1 0 1 2 3 4 5 6 7 8 9 20 Bleeding Score Likelihood ratio for VWD based on Vicenza bleeding assessment tool Elsevier 2007 CBC, PT, PTT. Fibrinogen or TT (optional) If bleeding history is strong consider performing initial VWD assays Prolonged PTT 1:1 mixing Corrected Initial VWD assay Not corrected Antiphospholipid antibody Other cause identified. eg. thrombocytopenia, Prolonged PT or TT Possible referral for appropriate evaluation FVIIF, IX and FXI Referral for appropriate evaluation VWF:Ag VWF:Rco FVIII Referral for specialized VWD studies Repeat VWD panel VWF:Rco/VWF:Ag Multimer analysis Collagen binding RIPA binding Plat VWF studies DNA sequencing Case Discussion Coagulation testing: PTT 42 sec (22.5-35 sec). Normal PT, and platelet count. Mixing studies: - PTT corrected with 1:1 mixing with Plasma Test F XI F IX:C F VIII:C vWF R:Co vWF:Ag Result 59% 78% 32% 35% 30% Normal 50-150% 60-150% 50-150% 45-200% 36-157% von Willebrand Disease ä Most common hereditary coagulation abnormality in human ä Estimated to occur in 1% to 0.1% of the population ä Female /Male ratio is 2:1 ä Borderline or modestly low VWF levels are unlikely to run in families, and usually symptomatic ä VWD subtype I and II are dominant. Type III is recessive ä Type II and III can be diagnosed by molecular genetic testing ä rVWF (BAX 111) has recently been introduced platelet F-VIII vWF endothelial cell Adherence of platelet to damaged endothelium is vWF dependent platelet plug fibrin clot Personal history of excessive bleeding Low VWF levels Known mutation consistent with VWD (type II) In absence of all three criteria, modestly low VWF levels should be treated as a risk factor for bleeding rather than a bleeding disorder GP Ib GP IIb& IIIa Decreased Increased binding binding to to platelet platelet VWD type 2A VWDtype type2M 2B VWD Complete deficiency Adhesion of VWF Impaired binding of site VWD type 3 VWF to collagen Reduced production GP Ia Decreased binding of vWF to factor VIII Aggregation VWD type 1site VWD type 2N VWD type 1C Collagen binding site F VIII N D1 D2 D3 GPIb GPIIb/IIIa Collagen A1 A2 A3 D4 C1 C2 C3 C Mutations 2A 2N 2B 2A 2M 2A Type 1 >100 different mutations throughout the gene, missense predominate Type 2A Mutation in proteolysis site at A2 region (most common) Type 2A Loss of propeptide, required for multimer formation from dimers Type 2A Mutation in C-terminus, required for dimer formation from monomers Type 2B Mutation in GPIb binding site, causing increased binding of vWF to GPIb Type 2M Mutation in GPIb binding site, causing decreased binding of vWF to GPIb Type 2N Mutation in N-terminis (FVIII binding) with decreased binding of vWF to factor VIII Type 3 Large null mutations Analysis of von Willebrand factor (VWF) multimers: The distribution of VWF multimers is analysed using sodium dodecyl sulphate (SDS)–agarose electrophoresis followed by immunostaining Local measure, pinching Topical antiseptic cream Nasal cauterization Topical hemostatic agents such as tranexamic acid Correcting underlying clotting problems (i.e. DDAVP)