Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Segmental Duplication on the Human Y Chromosome wikipedia , lookup
Gene therapy wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Human genetic variation wikipedia , lookup
Ridge (biology) wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Genetic engineering wikipedia , lookup
Primary transcript wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
DNA sequencing wikipedia , lookup
Genomic imprinting wikipedia , lookup
Microevolution wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Gene expression programming wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Epigenomics wikipedia , lookup
Copy-number variation wikipedia , lookup
Gene desert wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Transposable element wikipedia , lookup
History of genetic engineering wikipedia , lookup
Long non-coding RNA wikipedia , lookup
Oncogenomics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Designer baby wikipedia , lookup
Genome (book) wikipedia , lookup
Non-coding DNA wikipedia , lookup
Helitron (biology) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Public health genomics wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Human genome wikipedia , lookup
Minimal genome wikipedia , lookup
Genome editing wikipedia , lookup
Pathogenomics wikipedia , lookup
Genomic library wikipedia , lookup
Metagenomics wikipedia , lookup
Exome sequencing wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Human Genome Project wikipedia , lookup
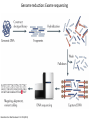
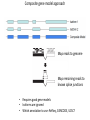
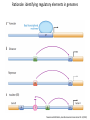
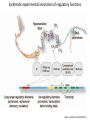
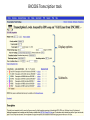
Genomics and High Throughput Sequencing Technologies: Applications Jim Noonan Department of Genetics Outline Personal genome sequencing •Rationale: understanding human disease •Variant discovery and interpretation •Genome reduction strategies (exome sequencing) Functional analysis of biological systems using sequencing •Transcriptome analysis: RNA-seq •Regulatory element discovery: ChIP-seq •Chromatin state profiling and the ‘histone code’ •Large-scale efforts: ENCODE and the NIH Epigenome Roadmap Whole genome sequencing: 1000 Genomes Nature 467:1061 (2010) The genetic architecture of human disease State, MW. Neuron 68:254 (2010) Challenge: Interpreting genetic variation Cooper and Shendure, Nat Rev Genet 12:628 (2011) Tools for identifying rare damaging mutations Protein-sequence based DNA-sequence based All humans have rare damaging mutations Damages protein Conserved Cooper and Shendure, Nat Rev Genet 12:628 (2011) Genome reduction: Exome sequencing Bamshad et al. Nat Rev Genet 12:745 (2011) Finding disease-causing rare variants by exome sequencing De novo mutation • • • • Screen unrelated trios for recurrence Likely to have functional effect Recurrence in independent affected individuals Absence in controls Reveal critical pathways in disease Sanders et al., Nature 485:237 (2012) Outline Personal genome sequencing •Rationale: understanding human disease •Variant discovery and interpretation •Genome reduction strategies (exome sequencing) •Challenges to de novo genome assembly using short reads Functional analysis of biological systems using sequencing •Transcriptome analysis: RNA-seq •Regulatory element discovery: ChIP-seq •Chromatin state profiling and the ‘histone code’ •Large-scale efforts: ENCODE and the NIH Epigenome Roadmap mRNA-seq workflow Martin and Wang Nat Rev Genet 12:671 (2011) Wang et al. Nat Rev Genet 10:57 (2009) Gene expression profiling by massively parallel RNA sequencing (RNA-seq) Mapping RNA-seq reads and quantifying transcripts Quantifying gene expression by RNA-seq Use existing gene annotation: • • • • • Align to genome plus annotated splices Depends on high-quality gene annotation Which annotation to use: RefSeq, GENCODE, UCSC? Isoform quantification? Identifying novel transcripts? Reference-guided alignments: • Align to genome sequence • Infer splice events from reads • Allows transcriptome analyses of genomes with poor gene annotation De novo transcript assembly: • Assemble transcripts directly from reads • Allows transcriptome analyses of species without reference genomes RNA-seq reads mapped to reference Normalization methods: Reads per kilobase of feature length per million mapped reads (RPKM) • • • What is a “feature?” What about genomes with poor genome annotation? What about species with no sequenced genome? For a detailed comparison of normalization methods, see Bullard et al. BMC Bioinformatics 11:94. What depth of sequencing is required to characterize a transcriptome? Wang et al. Nat Rev Genet 10:57 (2009) Considerations Gene length: • Long genes are detected before short genes Expression level: • High expressors are detected before low expressors Complexity of the transcriptome: • Tissues with many cell types require more sequencing Feature type • Composite gene models • Common isoforms • Rare isoforms Detection vs. quantification • Obtaining confident expression level estimates (e.g., “stable” RPKMs) requires greater coverage Pervasive alternative splicing in humans Wang et al. Nature 456:470 (2008) Composite gene model approach Map reads to genome Map remaining reads to known splice junctions • Requires good gene models • Isoforms are ignored • Which annotation to use: RefSeq, GENCODE, UCSC? Strategies for transcript assembly Garber et al. Nat Methods 8:469 (2011) ChIP-seq • Transcription factors • General transcription machinery • Modifications to histone tails • Methylated DNA Rationale: identifying regulatory elements in genomes Noonan and McCallion, Ann Rev Genomics Hum Genet 11:1 (2010) ChIP-seq peak calling ChIP-seq is an enrichment method Requires a statistical framework for determining the significance of enrichment ChIP-seq ‘peaks’ are regions of enriched read density relative to an input control Input = sonicated chromatin collected prior to immunoprecipitation There are many ChIP-seq peak calling methods Wilbanks and Facciotti PLoS ONE 5:e11471 (2010) The histone code Zhou et al. Nat Rev Genet 12:7 Mapping and analysis of chromatin state dynamics in nine human cell types Cell types: Marks: • H1 ESC • H3K4me3 (promoter/enhancer) • K562 (erythrocyte derived) • H3K4me2 (promoter/enhancer) • GM12878 (B-lymphoblastoid) • H3K4me1 (enhancer) • HepG2 (hepatocellular carcinoma) • H3K9ac (promoter/enhancer) • HUVEC (umbilical vein endothelium) • H3K27ac (promoter/enhancer) • HSMM (skeletal muscle myoblasts) • H3K36me3 (transcribed regions) • NHLF (lung fibroblast) • H4K20me1 (transcribed regions) • NHEK (epidermal keratinocytes) • H3K27me3 (Polycomb repression) • HMEC (mammary epithelium) • CTCF Ernst et al., Nature 473:43 (2011) Mapping and analysis of chromatin state dynamics in nine human cell types Ernst et al., Nature 473:43 (2011) Chromatin state dynamics at WLS Ernst et al., Nature 473:43 (2011) Functions associated with putative promoter and enhancer states • Annotation based on nearest TSS ChIP-seq: enhancer identification in vivo Visel et al. Nature 457:854 (2009) •p300 = enhancer-associated factor •p300 binding = ~90% predictive of enhancer activity Systematic experimental annotation of regulatory functions Myers, PLoS Biol 9:e1001046 (2011) The ENCODE Project http://genome.ucsc.edu/ENCODE/ The NIH Roadmap Epigenomics Project http://www.roadmapepigenomics.org/ ENCODE cell lines Myers, PLoS Biol 9:e1001046 (2011) ENCODE Project data access http://genome.ucsc.edu/ENCODE/ Genome Browser interface and data types Genome Viewer Categories of data: displayed as tracks Discrete intervals (genes) or continuous (transcription) Hyperlinks and pulldown tabs for individual tracks • • Go to track description page Hide or show data in genome viewer Some tracks include multiple datasets (‘subtracks’) •Go to track description page to select ENCODE Transcription track Display options Subtracks Conclusions Personal genomics is becoming a reality •Genome sequencing will be a routine diagnostic tool •$5,000 to sequence single genome; current cost for clinical resequencing of single genes •Your genome will be sequenced •Long-read sequencing will solve de novo assembly issues •Data analysis and interpretation RNA-seq and ChIP-seq •Identifying genes and annotating regulatory function within and among genomes •Computational issues: data normalization, peak calling, differential expression and binding •Large-scale studies revealing regulatory architecture of human & model genomes