Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
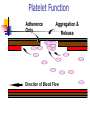
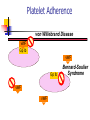
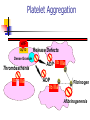
Thrombasthenia CPC 24th November 2002. Dr. Tariq Roshan Department of Hematology. Case History. SNS: a 3-year old Malay girl. Presented with 8 months H/O recurrent epistaxis. Failure to thrive. Problem started at the age of 1 year The condition became worse over the previous 8 months The frequency of epistaxis has been once in every 3-4 days Epistaxis was sometimes associated with gum bleeding Patient sought medical attention and was admitted to hospital Kota Bharu In HKB patient noted to be pale and diagnosed anemic secondary to chronic blood loss Patient was transfused and was investigated for the possible causes of bleeding including those of bleeding disorders No diagnosis was made during that admission. ENT examination in HKB showed no anatomical abnormalities 8 months later the patient was admitted with the worsening of her problem to HUSM Negative points in the history were Fever Easy bruising Petechiae Skin rashes Joint pain Family history Father and brother had history of epistaxis during their childhood which resolved spontaneously Physical examination GPE Height and weight below 3rd centile Pallor Cervical lymph nodes were palpable Generalized hypotonia with mild muscle wasting noted Systemic examination Examination of CNS, CVS and Respiratory system was normal Liver was palpable 5 cm below costal margin and spleen palpable 8 cm below costal margin Laboratory Investigations Patient ID Date APTT Cont. INR Mixing Test Cont. Plat. BT/CT Factor VIII Factor IX SNS 54.2 Sec. 33.9 Sec. 1.07 39.6 Sec. 32.4 Sec. Corrected 220 x109/L Prolonged 93.4% 38% Special Studies Both parents Factor IX was normal. vWF antigen assay HEMOSTASIS 182% Patient’s Haemoglobin was low and total white cell count was normal FBP showed microcytic hypochromic anemia Normal platelet count and morphology on light microscopy Laboratory Investigations contd. Patient’s RFTs were normal with mild elevation of ALT & ALP Blood sugar and urine analysis were also normal Patient was screened for TORCHES, all results were negative Sputum for AFB was negative Bone marrow aspiration and trephine biopsy were normal with no evidence of infiltration of abnormal cells. Adequate megakaryocytes Conclusions drawn from the Haemostasis data Patient ID Date APTT Cont. INR Mixing Test Cont. Plat. BT/CT Factor VIII Factor IX SNS 54.2 Sec. 33.9 Sec. 1.07 39.6 Sec. 32.4 Sec. Corrected 220 x109/L Prolonged 93.4% 38% Special Studies Both parents Factor IX was normal. vWF antigen assay HEMOSTASIS 182% The increased bleeding time and normal platelet count indicate a primary hemostasis function problem Such data may result from platelet disorders or vascular function disorders Haemophilia can be excluded considering the clinical presentation Low factor IX can be due to systemic disease or liver problem However, in view of normal INR, liver pathology as a possible cause of low factor IX, was excluded Low factor IX can also be seen in glycogen storage diseases as Gaucher disease and should be ruled out by investigation Hemostasis Primary vs. Secondary vs. Tertiary Primary Hemostasis Secondary Hemostasis Platelet Plug formation Dependent on normal platelet number & function Initial manifestation of clot formation Activation of the clotting cascade Deposition & Stabilization of fibrin Tertiary Hemostasis Dissolution of the fibrin clot Dependent on Plasminogen Activation Qualitative Defects in Primary Hemostasis Adhesion/Adherence Defects Glycoprotein (Gp) Ib deficiency Bernard-Soulier syndrome von Willebrand disease Aggregation defects Afibrinogenemia Release defect Acquired defects Antibodies to GP IIb-IIIa in NHL and HD Platelet Function Adherence Only Aggregation & Release Direction of Blood Flow Platelet Adherence von Willebrand Disease vWF Gp Ib vWF Gp Ib vWF vWF Bernard-Soulier Syndrome Platelet Release Function ADP Wiskott-Aldrich syndrome ß-thromboglobulin Platelet factor 4 Hermansky-Pudlak syndrome DenseGranule Platelet-derived Growth Factor Alpha-granule Gray platelet syndrome Fibrinogen Lysosome ChédiakHigashi anomaly Factor V Hydrolase Platelet Aggregation vWF Gp Ib Dense Granule Thrombasthenia IIb IIIa Release Defects ADP IIb IIIa D D ADP D IIb IIIa E Fibrinogen Afibrinogenemia Platelet Aggregation In-vivo platelet aggregation is induced by Thrombin mechanisms Stimulates ADP release Enhances formation of TxA2 Thromboxane A2, mediated through Arachidonic acid release form membrane phospholipid Cyclo-oxygenase Endoperoxidase Laboratory Tests for Primary Hemostasis Function Platelet count Bleeding time Platelet Aggregation Studies Luminance method Impedance method Clot retraction Not available Expected results: clots should normally be reduced by 50% of their original mass within 1 hour Flow cytometric studies for Glycoproteins Platelet Aggregometry % Transmittance Secondary Response Aggregation Plateau Shape Change Reagent Added Baseline Note: With impedance method there is increased resistance of the sample and ATP release Time Results of platelet aggregation studies No aggregation with arachidonic acid and ADP Markedly reduced aggregation with collagen Normal aggregation with ristocetin ATP release with thrombin is normal but absent with arachidonic acid Platelet Aggregometry interpretation Aggregating Agent Disorder ADP Epi Thr Ris Col Ara Bernard-Soulier syndrome Nor Nor Var Abn Var Nor Chédiak-Higashi anomaly Abn/Var Abn/Var Abn/Var Abn/Var Glanzmann thrombasthenia Abn Abn Granule release defects Abn Abn von Willebrand's disease Abn Nor Abn Abn Abn Flow cytometry for the detection of Gp defeciency The study of GP IIb IIIa showed reduction in PAC-1 which is a finding consistent with Glanzmann thrombasthenia However hepatosplenomegaly is not explainable by the diagnosis Patient was readmitted on 18-11-02 for further investigations to clarify the hepatosplenomegaly Thrombasthenia Synonyms: Glanzmann thrombasthenia, constitutional thrombopathy, hereditary hemorrhagic thrombopathy Background: Thrombasthenia was first describe in 1918 by Glanzmann when he noted purpuric bleeding in patients with normal platelet counts Typically, thrombasthenia is diagnosed at an early age Pathophysiology: Autosomal recessive trait The production and assembly of the platelet membrane glycoprotein IIb-IIIa is altered, preventing the aggregation of platelets and subsequent clot formation Review of platelet function Platelets adhere to the site of endothelial injury Activate Aggregate Secrete & promote further platelet recruitment & aggregation vWF binds to the exposed collagen and binds GP Ib-IX-V complex on the surface of platelet, adhering platelets to the site of injury Fibrinogen and vWF bind to the GP IIb-IIIa complex on the activated platelet’s surface, allowing cross-linking and formation of clot Specific Deficiency GP IIb and IIIa have separate genes on the long arm of chromosome 17 Specific genetic abnormalities of each GP include Missense mutations Nonsense mutations Splice site mutations Deletions and Point mutations Abnormalities in either gene or in the assembly of the complex result in an abnormal or deficient receptor Consequently One or other GP is not formed properly, leaving the other unpaired in the endoplasmic reticulum, where it is degraded Platelet aggregation is rendered deficient or completely absent Heterozygotes are asymptomatic Binding sites for thrombin are preserved in thrombasthenic platelets Patients are classified into: type1, type2, or the variant type, depending on the degree of GP IIb-IIIa deficiency, fibrinogen binding, and clot retraction Type 1 : most severe form, less than 5% of normal GP IIb-IIIa present with absent fibrinogen binding and clot retraction Type 2 : 10-20% of GP IIb-IIIa, normal to moderately deficient clot retraction with fibrinogen binding Variant type : 50% of the normal amount of GP IIbIIIa with extremely variable fibrinogen binding and clot retraction Frequency: Mortality/Morbidity: Death following bleeding approx. 5% Age: 300 cases are reported in medical literature 1st case diagnosed in HUSM Typically diagnosed during infancy Epistaxis is more severe in children but rare in adults History In the neonatal period: mucocutaneous bleeding In childhood: purpura, epistaxis & gingival bleeding The bleeding tendency decreases with age Additional presentations include GI bleeding Excessive bleeding at menarche & following parturition Post surgical bleeding Hemarthrosis and deep hematomas are unusual The absence of family history should not delay a workup for thrombasthenia as it is a recessive trait Beyond identification of hemorrhage, physical examination is usually of limited use Other disorders to be considered in the differential diagnosis: Gray platelet syndrome Hermansky-Pudlak syndrome Chediak-Higashi syndrome ITP DIC Medication-induced platelet inhibition Others Prostaglandin synthetase inhibitors (Aspirin, NSAIDS) ADP receptor inhibitors (Clopidogrel, Ticlopidine) Receptor blocking drugs (Dipyridamole) Beta-lactam antibiotics Heparin Alcohol Uremia hyperglobulinemias Lab investigations: Bleeding time Aggregation studies Platelet count and morphology PT/ APTT Flow cytometric studies Medical Care Emergency care: Refractory bleeding requires transfusion of normal platelets HLA-matched platelets is the treatment of choice In rare cases, antibodies to Gp IIb-IIIa are detectable Medical treatment: Antifibrinolytic agents inhibit fibrinolysis via inhibition of plasminogen activator substances Aminocaproic acid; may be useful in controlling bleeding after dental extraction Contraindicated in the evidence of active intravascular clotting process (DIC) Hematuria is relative contraindication Co-administration with estrogens may cause increase clotting factors, leading to hypercoagulable state Medical treatment contd Vasopressin analogs, act like ADH to increase factor VIII levels Desmopressin (DDAVP); synthetic vasopressin analog Clotting factors The use of DDAVP not recommended routinely Contraindicated in documented hypersensitivity Coagulation factor VIIa, recombinant Other therapies cited in literature IV Igs Repeated plasma pheresis Bone marrow transplant Conclusion & Take home message Final diagnosis of our patient was Glanzmann thrombasthenia Family studies for platelet function defect are recommended However, further investigations are required to clarify the cause of hepatosplenomegaly and low factor IX Refractory haemorrhage in the presence of normal platelet count and normal coagulation factors need to be investigated further (Now in HUSM) Thank you & Selamat Hari Raya