Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Community fingerprinting wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Western blot wikipedia , lookup
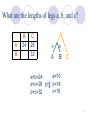
Two-hybrid screening wikipedia , lookup
Genetic code wikipedia , lookup
Biochemistry wikipedia , lookup
Point mutation wikipedia , lookup
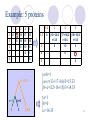
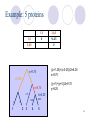
Construction of Phylogenetic Trees Walter M. Fitch and Emanuel Margoliash Science, New Series, Volume 155, Issue 3760(Jan. 20, 1967), 279-284 Speaker : Fang-Ling Lin Advisor : Prof. R.C. T. Lee National Chi-Nan University 1 Outline Basic nouns Construct phylogenetic tree Analyze the phylogenetic tree Reconstruction of the ancestral cytochrome c amino acid sequences. 2 Introduction Biochemists have attempted to use quantitative estimates of variance between substances obtained from different species to construct phylogenetic trees. These methods have not been completely satisfactory because 1. restricted 2. accuracy 3. mathematical 3 What is cytochrome c? Cytochrome c is a protein that participates in the metabolism of the mitochondrion . It will move from the mitochondrion to the cytoplasm and the cell will die. 4 Determining the Mutation Distance The mutation distance : The minimal number of nucleotides that would need to be altered in order for the gene for one cytochrome to code for the other. ACTGAT A C T G AT T C T - AT C TCTATC 5 Problem Given: Output: phylogenetic tree 6 The construction of the tree Assume there are proteins, A, B and C, and their mutation distances. A B B 24 C 28 32 There are two fundamental problems: 1. Which pair does one join together first? 2. What are the lengths of edges a, b, and c? 7 Which pair does one join together first ? It is simply by choosing the pair with the smallest mutation distance. A B B 24 C 28 32 A B C 8 What are the lengths of legs a, b, and c? A B B 24 C 28 32 a+b=24 a+c=28 b+c=32 c a b A B C a=10 b=14 c=18 9 When information from more than three proteins is utilized When information from more than three proteins is utilized, the basic procedure is the same. One then simply joins two subsets to create a single subset. Until all proteins are members of a single subset. 10 Example: 5 proteins 1 1,2 1 2 3 4 5 0 1 13 17 16 0 12 16 15 0 10 8 3 0 1 4 2 3 4 5 0 c=14.33 a=1 b=0 1 2 3,4,5 1,2 0 3 4 5 (13+12)/2 (17+16)/2 (16+15)/2 =12.5 =16.5 =15.5 0 10 8 0 1 5 0 a+b=1 a+c=(13+17+16)/3=15.33 b+c=(12+16+15)/3=14.33 a=1 b=0 c=14.33 11 Example: 5 proteins 1,2 1,2 3 4,5 0 12.5 (16.5+15.5)/2 =16 0 (10+8)/2 =9 3 1 1 0 2 , 3 4,5 0 c=12 a+b=1 a+c=(16.5+10)/2=13.25 b+c=(15.5+8)/2=11.75 a=1.25 4 b=-0.25 5 a=1.25 b=-0.25 c=12 12 Example: 5 proteins 1,2 1,2 3,4,5 0 (12.5+16)/2 =14.25 3,4,5 0 a+b=9 a+c=12.5 b+c=16 c=9.75 b=6.25 a=2.75 1 1 1.25 0 2 3 a=2.75 b=6.25 c=9.75 4 -0.25 5 13 Example: 5 proteins 1,2 1,2 3,4,5 0 14.25 3,4,5 0 ((x+1.25)+(x-0.25))/2=6.25 x=5.75 c=9.75 y=9.25 x=5.75 2.75 1 1 b=6.25 1.25 0 2 3 ((y+1)+(y+0))/2=9.75 y=9.25 4 -0.25 5 14 Testing Alternative Trees In this method, the output is generated by input, and the results are the same by using the same input every time. Since a particular assignment of species to A and B subsets defines a tree, thus different assignments of species to A and B produce different trees. Check this out. Fig. 1 is the best of 40 phylogenetic trees. 15 Phylogenetic Tree of 20 species •Back 1 •Back 2 Fig.1 16 Reconstructed distances j reconstruct value i original input Values in the upper right half of the table are reconstructed distances found by summing the leg lengths in Fig.1. 17 Standard deviation the percentage of change from the input data standard deviation: summed over all values of i<j 18 The statistically optimal tree In testing phylogenetic alternatives, one is seeking to minimize the percent “standard deviation.” Fig.1 has a percent “standard deviation” of 8.7, the lowest of the 40 alternatives so far tested. The percent “standard deviation” for the initial tree was 12.3. 19 The statistically optimal tree 20 Fig.1 is remarkably like that constructed in accord with classical zoological comparisons. Almost all the alternative phylogenetic schemes tested involved rearrangements with the groups birds (turkey, chicken) and nonprimate mammals (cow, sheep, pig). 21 Three noticeable deviations Birds of flight (Neognathae) and penguin (Impennae) Kangaroo v.s. nonprimate mammals and placental mammals v.s. marsupials The turtle appears more closely associated with the birds than to its fellow reptile the rattlesnake. Fig.1 22 Indeed, from any phylogenetic ancestor, today’s descendants are equidistant with respect to time but not equidistant genetically. The method indicates those lines in which the gene has undergone the more rapid changes. For example, The mutation distance between mammals and primates is 7.5 and that between mammals and non-primates is 5.8. The change in the cytochrome c gene has been much more rapid in the descent of the primates than in that of the other mammals. Fig.1 23 Reconstruction of the ancestral cytochrome c amino acid sequences. The procedure is dependent upon the phylogenetic tree on which these sequence data are arranged. 24 Amino acid No. Ancestral Mammal Ancestral Primate Monkey Man ---------Kangaroo ---------Rabbit ---------Dog Ancestral Ungulate Pig Ancestral Perissodactyl Donkey Horse 17 18 21 39 41 50 52 53 56 64 66 68 89 94 95 98 109 V Q L H U P O S A E Y A L W M W M W M V Q V Q V Q V Q V Q V Q S S S L L L L L L H H H H N H H H H U U U U W U U U U P P P P P P V P P O O F F F F F O F S S S S S S S S S L L L A A A A A A E E W E W E E E E Y A Y A Y A Y A Y A Y A Y A Y G Y G V V V L L L L L L I G L N I I I I I I I I E G G G Y Y Y A Y Y L L L L L L L L L N N N N N N N N I V Q L H U P F S A E Y G L I Y Q N V Q L H U P F S A E Y G L I Y Q N V Q L H U P F S A E I G L I E Q N V Q L H U P F S A E I G L I E Q N V Q L H U P F E A E I G L I E Q N 25 There is presently no detectable relationship between the primary structures of cytochrome c and those of hemoglobins. The reconstruction and comparison of the ancestral amino acid sequences may reval a homology that cannot be detected in present-day proteins. The employment of such ancestral sequences may be generally useful for detecting common ancestry not otherwise observable. 26 Thank you ! 27