Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Drug design wikipedia , lookup
Gene expression wikipedia , lookup
Magnesium transporter wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Point mutation wikipedia , lookup
Metabolomics wikipedia , lookup
Interactome wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Genetic code wikipedia , lookup
Protein purification wikipedia , lookup
Biosynthesis wikipedia , lookup
Western blot wikipedia , lookup
Metalloprotein wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Structural alignment wikipedia , lookup
Two-hybrid screening wikipedia , lookup
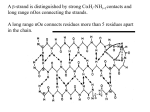
Protein NMR terminology COSY- Correlation spectroscopy Gives experimental details of interaction between hydrogens connected via a covalent bond H H H H C C C N NOESY- Nuclear Overhauser effect spectroscopy Gives peaks between pairs of hydrogen atoms near in space (1.5-5 Å) (and not necessarily sequence) CH H H N Fingerprint region 1 2 dH TOCSY Walk along the sequence gH bH 3 4 5 COSY 9.0 NH O C 7.0 NOE CH3 N C C H H O NOE COO CH2 H H O N C C CH2 Ala CH H3C - CH3 N H H 7.0 Connectivites by NOE dN - Connects CH of residue i to NH of i+1 dbN - Connects CbH of resdiue i to NH of i+1 dNN - Connects NH of residue i to NH of i+1 LEU dN H dN dNN N dbN H ALA VAL C dNN C H3C H N CH3 CH d O bN C C H N C H CH3 dNN O H dN CH2 dbN Hi-NHi+3 Hi-NHi+1 i+3 H i+2 An -helix can be recognised by repeating patterns of short range nOes. A short range nOe is defined as a contact between residues less than five apart in N i+4 the sequence (sequential nOes H connect neighbouring residues) NOE H For an -helix we see Hi-NHi+3 and Hi-NHi+4 nOes. i Assignment of secondary structural segments • sequential stretches of residues with consistent secondary structure characteristics provide a reliable indication of the location of these structural segments A b-strand is distinguished by strong CHi-NHi+1contacts and long range nOes connecting the strands. A long range nOe connects residues more than 5 residues apart in the chain. A real example. The rat fatty acid acyl carrier protein. Involved in fatty acid biosynthesis and part of a larger subunit, the synthase, Is it structured by itself?? Summary of the Sequential and Secondary NOEs observed for rat FAS ACP - most definitely structured NHi-NHi+1 iNHi+1 biNHi+1 GDGEAQRDLVKAVAHILGIRDLAGINLDSSLADLGLDSLMGVEVR D D D DD D D D NHi-NHi+2 Hi-NHi+2 Hi-NHi+3 Hi-NHi+4 Hi-bHi+3 CSI J 0-00000---------+-0-0--0+--+0+---+00+-0-----+ ++ -------+--+++++ +++-+++ --- ----- --QILEREHDLVLPIREVRQLTLRKLQEMSSKAGSDTELAAPKSKN NHi-NHi+1 iNHi+1 biNHi+1 D D D D DD D D DDD NHi-NHi+2 Hi-NHi+2 Hi-NHi+3 Hi-NHi+4 Hi-bHi+3 CSI J -----0+-+0++--0--00+--------00000000+0+00-00 -++-+++ - -- -+-+ - -+- +++++++ +++ So I have assigned the NMR spectrum and connected the amino acids. I have a good idea of the secondary structure. What next?? At this point we notice there are still many nOes we have not assigned on the 2D spectrum. These are neither sequential or short range. They are long-range and connect residues more more than 5 amino acids apart (But still close in space!). O H N CH C O H N CH 2 Asn C O CH C Gly H N H H OH Identified as an asparagine aminohydrogen from COSY spectra HO O C CH 2 Glu H2C C O CH NH 2 NOE indicated the asparagine amino-hydrogen is near a glutamate acidic hydrogen Schematic showing long range nOes in the lac headpiece protein What next? STRUCTURE CALCULATIONS •From NOE I know close atom-atom distances, but that doesn’t give a structure •The information you have up to this stage is a list of distance constraints •The structure can be determined by inputting this information to computer minimization software. •The computer program also contains information about amino acids, bond lengths/angles and standard information about atom-atom interactions such as minimum distance (i.e. Van der Waals radii) •With all this information you can generate a model of the structure. Important: NMR gives you a number of possible solutions (all almost identical, rmsd <1Å), This can range from 5-20 models X-ray crystallography give one average structure NMR structures can be averaged to give one average structure as well Excerpt from an NOE table for Actinorhodin Polyketide ACP - 1997 This file contained ~ 700 lines of nOe restraints ! Thr7 NH assign (resid 7 and name HN )(resid 75 and name HD1* ) 4.0 2.2 0.5 assign (resid 7 and name HN )(resid 75 and name HD2* ) 4.0 2.2 0.5 assign (resid 10 and name HN )(resid 75 and name HD2* ) 5.0 3.2 0.5 assign (resid 10 and name HN )(resid 75 and name HD1* ) 3.3 1.5 1.0 assign (resid 72 and name HN )(resid 31 and name HD1* ) 5.0 5.0 0.5 assign (resid 72 and name HN )(resid 31 and name HD2* ) 3.3 1.5 0.5 assign (resid 72 and name HN )(resid 31 and name HB* ) 4.0 4.0 1.5 assign (resid 72 and name HN )(resid 31 and name HA ) 4.0 4.0 1.0 75 and name HN )(resid 10 and name HD1* ) 4.5 4.5 1.0 ! Leu10 NH !Arg72 NH ! Leu 75 NH assign (resid The simulated annealing protocol - begin by simulating a 1000K heat bath and generate an extended model strand Start Apply the distance restraints from the NOE data (perhaps 1000 restraints for a protein of 90 amino acids). Weight the nOes to favour the formation of local secondary structure and later long range structure. Allow chain to move through itself 30 ps Start to cool the system and increase the penalty for bad contacts. 20 ps Minimize the final structure to see if it satisfies all the nOes A simulated annealing trajectory over the first few picoseconds 4 helices begin to ‘condense’ Unfolded Correctly folded Challenges for Interpreting 3D Structures • To correctly represent a structure (not a model), the uncertainty in each atomic coordinate must be shown • Polypeptides are dynamic and therefore occupy more than one conformation – Which is the biologically relevant one? Representation of Structure Conformational Ensemble Neither crystal nor solution structures can be properly represented by a single conformation Intrinsic motions Imperfect data Uncertainty RMSD of the ensemble Representations of 3D Structures C N These 2D methods work for proteins up to about 100 amino acids, and even here, anything from 50-100 amino acids is difficult. We need to reduce the complexity of these 2D spectra. 1 16 1 H O HN 12 12 C 14 N R2 14 N C 16 O 12 C 12 C 1 R1 1 H HN We can start by replacing 14N with 15N, a spin 1/2 nucleus. Run a ‘COSY’ type experiment that correlates an amide proton with the 15N nuclei. This is a heteronuclear experiment, I.e. we are looking at two different nuclei, a 1H and a 15N nucleus. The ‘COSY’ type experiment is beyond the scope of these lectures but is known as HSQC, or heteronuclear single quantum coherence spectroscopy. This refers to how the magnetisation is transferred from the 1H to the 15N. So how well dispersed are the 15N shifts? Is it worth trying to separate our spectra out based on their differences? 1H-15N HSQC of rat FAS ACP Why? •The more we understand about a protein and its function, the more we can do with it. It can be used for a new specific purpose or even be redesigned too carry out new useful functions (biotechnology & industry). •We can use this knowledge to help understand the basis of diseases and to design new drugs (medicine & drug design). •The more knowledge we have how proteins behave in general, the more we can apply it to others (protein families etc) A case study - Leukocyte function associated protein-1 (LFA-1) This protein is involved in tethering a leukocyte to a endothelium, allowing migration through the tissue to a site of inflammation. One domain of LFA-1, the I-domain is 181 amino acids and undergoes a conformational change where helix 7 slides down the protein, switching it into an active open form. This open form is competent for cell surface binding. If we can stop this switch, we may have an anti-inflammatory mechanism Inflammation (chronic) is responsible for asthma and arthritis. Developed small molecule inhibitors and test binding O- O O N S A B C N N D N O Weak binding mM to mM see a migration of the peaks A more successful inhibitor- nM ‘tight’ binding. See unbound and bound populations Solve NMR structure of complex… Helix 7 is prevented from shifting NMR is a diverse tool with which we can study protein structure. It gives us information in solution under ‘physiological’ conditions 2D and 3D techniques combined with modern assignment methods have allowed proteins up to 40 kDa to be solved. The power of NMR lies not just with its ability to solve structures but also its ability to probe binding of ligands and partner proteins in ‘real’ time. Many aspects we have not had time to deal with. NMR reveals how proteins move in solution - can see domains flexing with different timescale motions. These often correlate with binding patches on the protein. Textbook I recommend reading. J Evans - Biomolecular NMR Spectroscopy. Chapter 4. Protein Structure, pages 147-174. After p174 numerous examples of NMR structures, labelling etc. Chapter 2. More high level NMR approach - description of how pulse sequences (I.e. COSY, TOCSY, HNCA etc) work. Beyond the scope of the course but may be of interest. Chapter 3. Details of calculations - for you details not important but will give you more of an idea of how we use the NMR data to calculate the structure.