Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Biochemistry wikipedia , lookup
Paracrine signalling wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Drug design wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Gene expression wikipedia , lookup
Magnesium transporter wikipedia , lookup
Expression vector wikipedia , lookup
Metalloprotein wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Interactome wikipedia , lookup
Protein purification wikipedia , lookup
Western blot wikipedia , lookup
Two-hybrid screening wikipedia , lookup
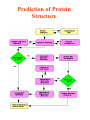
Molecular modelling / structure prediction (A computational approach to protein structure) Thomas Huber Department of Mathematics Room 724, Priestley building [email protected] Today: • Why bother about proteins/prediction • Concepts of molecular modelling – The physicist’s approach – The biologist’s approach • Get a feel for usefulness/uselessness • Where is the future going? Why do we care about Protein Structures/ Prediction? • Academic curiosity? – Understanding how nature works • Drug & Ligand design – Need protein structure to design molecules which inhibit/excite • cure all sorts of diseases • Protein design – making better proteins • sensor proteins • industrial catalysts (washing powder, synthetic reactions, …) • Urgency of prediction – 104 structures are determined • insignificant compared to all proteins – sequencing = fast & cheap – structure determination = hard & expensive Three basic choices in molecular modelling • Representation – Which degrees of freedom are treated explicitly • Scoring – Which scoring function (force field) • Searching – Which method to search or sample conformational space The physicist’s approach: Folding by 1st principles Concept: Doing what nature does • Representation: atomic level • Scoring: physical force field • Searching: Newton’s equations of motion Naïve idea? • Levinthal’s paradox (1968) – 3 possible rotamers per dihedral angle astronomical number of conformations • Golf course scenario Levinthal’s paradox is irrelevant • Folding is not a random process Bumpy bowl scenario Why are folding simulations still unsuccessful? • • • • Simulations computational expensive Force fields are not good Gross approximations in simulations Nature uses tricks • Posttranslational processing • Chaperones • Environment change Is a physical approach useless? • No! • Very useful aid to structure determination / refinement – Experimentally observed structural data very incomplete • NMR: only distances < 6Å • Xtallography: only 50% of data can be measured (phase information missing) – Physico-chemical information and complement experimental data • Give dynamical picture of structure Biologist’s approach: Prediction by induction Concept: Homologous sequences fold into similar structures • Representation: amino acid sequence • Scoring: sequence similarity (identity) • Searching: optimal string matching (with gaps and insertions) Validation of concept (Rost, 1999) • >106 sequence alignments between protein pairs • Optimal discrimination between similar and dis-similar structure Is it useful? • PDB statistics: – 104 protein structures determined – <103 protein folds Template recognition Alignment Alignment correction Backbone generation Loop building Side chain generation Overall model refinement Model verification Force field • • • • • • • • Sequence score 8 Modelling steps – Comparison with Experimental results – Steric overlap – Ramachandran plot Limiting factors How good are homology models? • G.V. Vried 1998: 34 homologous protein pairs What about side chains? • Biology happens in side chains • Packing side chains in protein core is not a trivial problem – Many alternative arrangements – High energy barriers Accuracy of modelled side chains • Dunbrack SCWRL results – 299 monomeric proteins – 40263 side chains The Next Step: Computational Proteomics • Mass scale homology modelling of entire genomes – Lots of sequence data – First pick the easy cases – Computers are cheap and work 7-24 Prediction of Protein Structure How to detect remote homologues • Fold recognition using threading – Combine concepts of physicist and biologists • Predicting secondary structure • More about that in BIOL3004 – Structural biology elective • Tue 8/5 10am • Thu 10/5 10am – Database mining elective • L10 Take home messages • Computational approaches are – Not perfect – Yet indispensable • Molecular modelling has huge potential in structural biology – Currently 104 structures in PDB – For every sequence in the Swissprot database with homology to a structure in the PDB models are available!! – Vast amount of data still to come • Levinthal paradox – Is true – BUT not relevant • Different aims need different approaches (3 choices of MM!) – modelling enzyme reactions – modelling protein folding – weather forecast Clever approaches more important than bigger computers