Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
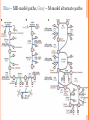
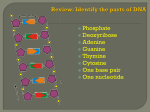
IN SILICO METHOD FOR MODELING METABOLISM AND GENE PRODUCT EXPRESSION AT GENOME SCALE Lerman, Joshua A., Palsson, Bernhard O. Nat Commun 2012/07/03 SO FAR – METABOLIC MODELS (M-MODELS) Predict reaction flux Genes are either ON or OFF Special ‘tricks’ to incorporate GE (iMAT) ‘tricks’ are imprecise, more tricks needed (MTA) Objective function debatable Usually very large solution space Flux loops are possible leading to unrealistic solutions. No regulation incorporated NEW – METABOLISM AND EXPRESSION (ME-MODELS) Add transcription and translation Account for RNA generation and degradation Account for peptide creation and degradation Gene expression and gene products explicitly modeled and predicted All M-model features included GE and proteomic data easily incorporated No regulation incorporated. ME-MODEL: THE DETAILS THE CREATURE Model of the hyperthermophilic Thermotoga maritime (55-90 °C) Compact 1.8-Mb genome Lots of proteome data Few transcription factors Few regulatory states… ADDING TRANSCRIPTION AND TRANSLATION TO MODEL MODELING TRANSCRIPTION (DECAY AND DILUTION OF M/T/R-RNA) Flux creating mRNA: 𝑉𝑡𝑟𝑎𝑛𝑠𝑐𝑟𝑖𝑝𝑡𝑖𝑜𝑛 Fluxes deleting mRNA: 𝑚𝑅𝑁𝐴 𝑆𝑒𝑐 (GE) 𝑉𝑚𝑅𝑁𝐴𝑑𝑖𝑙𝑢𝑡𝑖𝑜𝑛 (mRNA transferred to daughter cell) 𝑉𝑚𝑅𝑁𝐴𝑑𝑒𝑔𝑟𝑎𝑑𝑎𝑡𝑖𝑜𝑛 (NTPNMP) Controlled by two coupling constants: 𝜏𝑚𝑅𝑁𝐴 (mRNA half life, from lab measurements) 𝑇𝑑𝑜𝑢𝑏𝑙𝑖𝑛𝑔 = ln 2 growth rate (lab measured or sampling) 𝜏𝑚𝑅𝑁𝐴 𝑉𝑑𝑒𝑔𝑟𝑎𝑑𝑎𝑡𝑖𝑜𝑛 𝑇𝑑 𝑇𝑑 removed for every 𝜏𝑚𝑅𝑁𝐴 Fluxes are coupled: 𝑉𝑑𝑖𝑙𝑢𝑡𝑖𝑜𝑛 ≥ Means 1 mRNA must be times it is degraded Cell spends energy in rebuilding NMPNTP MODELING TRANSLATION: MRNAENZYMES Flux creating peptides: 𝑉𝑡𝑟𝑎𝑛𝑠𝑙𝑎𝑡𝑖𝑜𝑛 Translation limited by 𝑘𝑡𝑟𝑎𝑛𝑠 𝑃𝑟𝑜𝑡 𝑆𝑒𝑐 𝑚𝑅𝑁𝐴 𝑃𝑟𝑜𝑡∙𝑆𝑒𝑐 , upper bound on rate of single mRNA translation, estimated from protein length, ribosome translation-frame and tRNA linking rate (global) Fluxes are coupled: 𝑉𝑑𝑒𝑔𝑟𝑎𝑑𝑎𝑡𝑖𝑜𝑛 𝑉𝑡𝑟𝑎𝑛𝑠𝑙𝑎𝑡𝑖𝑜𝑛 ≥ 𝐾𝑡𝑟𝑎𝑛𝑠 ∙ 𝜏𝑚𝑅𝑁𝐴 Means 1 mRNA must be degraded every 𝐾𝑡𝑟𝑎𝑛𝑠 ∙ 𝜏𝑚𝑅𝑁𝐴 times it is translated MODELING REACTION CATALYSIS 𝑉𝑐𝑎𝑡𝑎𝑙𝑦𝑠𝑖𝑠 = 𝑘𝑐𝑎𝑡 𝑘𝑐𝑎𝑡 𝐸 𝑆 Κ𝑀 + 𝑆 (Michaelis-Menten kinetics) 𝑆𝑢𝑏𝑠𝑡𝑟𝑀𝑜𝑙𝑒𝑐𝑢𝑙𝑒 𝑃𝑟𝑜𝑑𝑀𝑜𝑙𝑒𝑐𝑢𝑙𝑒∙𝐶𝑜𝑚𝑝𝑒𝑥∙𝑆𝑒𝑐 is turnover number 𝐸 is complex concentration [𝑆] is substrate concentration Κ 𝑀 is substrate-catalyst affinity Assume 𝑉𝐶𝑜𝑚𝑝𝑙𝑒𝑥𝐷𝑒𝑔𝑟𝑎𝑑𝑎𝑡𝑖𝑜𝑛 ≪ 𝑉𝑐𝑜𝑚𝑝𝑙𝑒𝑥𝐷𝑖𝑙𝑢𝑡𝑖𝑜𝑛 𝑉𝑐𝑎𝑡𝑎𝑙𝑦𝑠𝑖𝑠 𝑉𝐷𝑖𝑙𝑢𝑡𝑖𝑜𝑛 ≥ 𝑘𝑐𝑎𝑡 ∙ 𝑇𝑑 Means one complex must be removed for every 𝑘𝑐𝑎𝑡 ∙ 𝑇𝑑 times it catalyzes Whole proteome synthesized for doubling Fast catalysis faster doubling (dilution) BUILDING THE OPTIMIZATION FRAMEWORK M-MODEL - REMINDER Total Biomass Reaction: Experimentally measure lipid, nucleotide, AA, growth and maintenance ATP Integrate with organism 𝑇𝑑 to define reaction approximating dilution during cell formation Cellular composition known to vary with 𝑇𝑑 Cellular composition known to vary with media LP used to find max growth subject to (measured) uptake rates ME-MODEL Structural Biomass Reaction: Account only for “constant” cell structure Cofactors like Coenzyme A DNA like dCTP, dGTP Cell wall lipids Energy necessary to create and maintain them Model approximates a cell whose composition is a function of environment and growth rate Cellular composition (mRNA, tRNA, ribosomes) taken into account as dynamic reactions LP used to identify the minimum ribosome production rate required to support an experimentally determined growth rate VALIDATION RNA-TO-PROTEIN MASS RATIO RNA-to-protein mass ratio (r) observed to increase as a function of growth rate (μ) Emulate range of growths in minimal medium Use FBA with LP to identify minimum ribosome production rate required to support a given μ Assumption: expect a successful organism to produce the minimal amount of ribosomes required to support expression of the proteome Consistent with experimental observations, MEModel simulated increase in r with increasing μ COMPARISON TO M-MODEL max biomass on minimal media, many solutions −5 chance of Sample and approx. Gaussian, 10 finding solution as efficient as ME-model. Can be found by minimizing total flux (many solutions stem from internal flux loops). OPTIMAL PATHWAYS IN ME-MODEL Produces small metabolites as by-products of GE Accounts for material and energy turnover costs Includes recycling S-adenosylhomocysteine, (by-product of rRNA and tRNA methylation) and guanine, (by-product of tRNA modification) Frugal with central metabolic reactions, proposes glycolytic pathway during efficient growth M-Model indicates that alternate pathways are as efficient Blue – ME-model paths, Gray – M-model alternate paths SYSTEM LEVEL MOLECULAR PHENOTYPES Constrain model to μ during log-phase growth in maltose minimal medium at 80 °C Compare model predictions to substrate consumption, product secretion, AA composition, transcriptome and proteome measurements. Model accurately predicted maltose consumption and acetate and H2 secretion Predicted AA incorporation was linearly correlated (significantly) with measured AA composition DRIVING DISCOVERY Compute GE profiles for growth on medium: L-Arabinose/cellobiose as sole carbon source Identify conditionally expressed (CE) genes essential for growth with each carbon source In-vivo measurements corroborate genes found in simulation – evidence of tanscript. regulation CE genes may be regulated by the same TF Scan promoter and upstream regions of CE genes to identify potential TF-binding motifs Found high-scoring motif for L-Arab CE genes and a high-scoring motif for cellobiose CE genes L-Arab motif similar to Bacillus subtilis AraR motif SUMMARY ADVANTAGES Because ME-Models explicitly represent GE, directly investigating omics data in the context of the whole is now feasible For example, a set of genes highly expressed in silico but not expressed in vivo may indicate the presence of transcriptional regulation Discovery of new TF highlights how ME-Model simulations can guide discovery of new regulons DOWNSIDES ME-model is more intricate then M-model, more room for unknown/incomplete knowledge May keep ME-model simulations far from reality on most organisms Lack of specific translation efficacy for each protein Lack of specific degradation rates for each mRNA lack of signaling Lack of regulatory circuitry THANK YOU