Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Epigenetics of neurodegenerative diseases wikipedia , lookup
Epitranscriptome wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Microevolution wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Designer baby wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
DNA vaccination wikipedia , lookup
History of genetic engineering wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Protein moonlighting wikipedia , lookup
Transfer RNA wikipedia , lookup
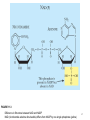
Genome editing wikipedia , lookup
Helitron (biology) wikipedia , lookup
Genetic code wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Chapter 11 Protein Engineering Introduction Engineering Disulfide Bonds improving Stability in Other Ways Changing Binding Site Specificity Structural Scaffolds Directed Evolution Adding New Functional Groups Using Nonnatural Amino Acids Recombinant Domains DNA Shuffling Combinatorial Protein Libraries Biomaterials Design Relies on Protein Engineering Engineered Binding Proteins Overlapping-extension PCR 2 3 4 5 6 7 Increasing the range of industrial enzymes has three facets. First, modem biology has identified many novel enzymecatalyzed reactions that may be of industrial use. Second, it is now possible to produce desired proteins in large amounts because of gene cloning and expression systems. Third, the sequence of the protein itself may be altered by genetic engineering to improve its properties. This is known as protein engineering. 8 ENGIINEERING DISUFIDE BONDS Disulfide bonds are especially important for those proteins found outside the cell in oxidizing environments. In practice, most enzymes used industrially will be exposed to such oxidizing conditions, and therefore disulfide bonds are particularly relevant. Introduction of extra disulfide bonds is a relatively straightforward way to increase the stability of proteins. The first step is to simply introduce two cysteine residues into the polypeptide chain. Then, under oxidizing conditions, these will form a disulfide bond provided that the polypeptide chain folds so as to bring the two cysteines into dose contact. Obviously, for this approach to work, the tertiary structure of the protein must be known so that the cysteines can be inserted in appropriate positions (Fig. 11.1). In general, the longer the loop of amino acids between the two cysteines, the greater the increase in stability. Note however, that formation of a disulfide linkage can create a strained conformation if the two cysteines are not properly aligned. This may result in a decrease in stability of the protein. 9 FIGURE 11.1 Introduction of Disulfide Bonds A disulfide bond can be added to a protein by changing two amino acids into cysteines by site-directed mutagenesis. When the engineered protein is put under oxidizing conditions, the two cysteines form a 10 disulfide bond, holding the protein together at that site. This approach has been demonstrated using the bacterial virus T4. lysozyme from The polypeptide chain of 164 amino acids folds into two domains and has two cysteines, neither involved in disulfide bond formation in the wild-type protein. One of these, Cys54, was first mutated to Thr to avoid formation of incorrect disulfides. The other, Cys97, was retained for use in disulfide formation. Extensive analysis of possible locations for disulfides was carried out. Those disulfides that might impair other stabilizing interactions in the protein were eliminated. This left three possible disulfide bonds that should theoretically promote stability, located between positions 3 and 97, 9. and 164, and 21 and 142 (Fig. 11.2). 11 FIGURE 11.2 Disulfide Engineering of T4 Lysozyme T4 lysozyme has two domains. The N-terminal region is shown in green and red, and the C-terminal region is in blue. These are linked by an alpha helix (purple). Disulfide bonds were added at three locations in T4 lysozyme to increase the stability. The first disulfide was between positions 9 and 164. This links the first alpha helix at the N terminus with the C-terminal tail. The second disulfide is between positions 2 and 97, which links the N- and C-terminal domains. Finally, the third disulfide links position 21 in the N-terminal domain with 142 in the C-terminal domain. The figure depicts alpha helices as barrel-shaped and beta-sheets as green arrows. 12 To test these experimentally, five amino adds (Ile3, Ile9, Thr21, Thr142, and Leu164) were converted to Cys in various combinations. Stability was measured by thermal denaturation; the melting temperature, Tm, is the temperature at which 50% of the protein is denatured. 13 IMPROVING STABILITY IN OTHER WAYS Because of the effects of entropy, the greater the number of possible unfolded conformations, the more likely a protein is to unfold. Decreasing the number of possible unfolded conformations therefore promotes stability. disulfide linkages. Glycine (-) proline (+) hydrophobic residues 14 Because of its asymmetrical structure, the alpha helix is actually a dipole with a slight positive charge at its N-terminal end and a slight negative charge at its Cterminal end. The presence of amino add residues with the corresponding opposite charge dose to the ends of an alpha helix promotes stability. In natural proteins the majority of alpha helixes are stabilized in this manner. However, in cases where such stabilizing residues are absent, protein engineering may create them. Asparagine and glutamine residues are relatively unstable. High temperature or extremes of pH convert these amides to their corresponding adds, aspartic add and glutamic add. The replacement of the neutral amide by the negatively charged carboxyl may damage the structure or activity of the protein. This may be avoided by engineering proteins to replace Asn or Gln by an uncharged hydrophilic residue of comparable size, such as Thr. 15 CHANGING BINDING SITE SPECIFICITY In addition to altering the overall stability of a protein, it is possible to deliberately change the active site. The most straightforward alterations to make are those that change the binding specificity for the substrate enzyme mechanism. or a cofactor, but do not disrupt the Changing the specificity for a cofactor or substrate may be useful, either to make the product of the enzyme reaction less costly or to change it chemically. This principle has been demonstrated with several enzymes that use the cofactors NAD or NADP to carry out dehydrogenation reactions. NAD is used by dehydrogenases in degradative pathways, and the respiratory chain oxidizes the resulting NADH. In contrast, biosynthetic enzymes use NADP. Structurally Generally, they differ only in NADP having an extra phosphate group attached to the ribose ring (Fig. 11.3). This gives NADP an extra negative charge and, not surprisingly, enzymes that prefer NADP have somewhat larger binding pockets with positively charged amino acid residues at the bottom. Enzymes that favor NADH often have a negatively charged amino acid residue in the corresponding position. 16 FIGURE 11.3 Difference in Structure between NAD and NADP NAD (nicotinamide adenine dinucleotide) differs from NADP by one single phosphate (yellow) 17 Several enzymes that use NAD or NADP have been engineered to change their reference For lactate dehydrogenase (LDH) of most bacteria uses reduced NAD, not NADP, to convert pyruvate to lactate. A conserved aspartate provides the negative charge at the bottom of the cofactor binding pocket that excludes NADP. If this is changed to a neutral residue, such as serine, the enzyme becomes able to use both NAD and NADP. If, in addition, a nearby hydrophobic residue in the cofactor pocket is replaced by a positively charged amino arginine), the enzyme now prefers NADP to NAD (Fig. 11.4). acid (such as lysine or 18 FIGURE 11.4 Changing Cofactor Preference of Lactate Dehydrogenase Lactate dehydrogenase (LDH) preferentially binds NAD because the binding pocket has an aspartic acid. The negatively charged carboxyl repels the negatively charged phosphate of NADP. Changing the aspartic acid to serine allows either NAD or NADP bind to LDH. Adding a positively charged lysine 19 makes the pocket more attractive to the NADP. LDH for its substrate The specificity of can be altered in a similar way. The natural substrate lactate is a three carbon hydroxyacid. It is possible to alter several residues surrounding the substrate binding site without impairing the enzyme reaction mechanism. By replacing a pair of alanines with glycines, the binding site can be made larger. By replacing hydrophlic residues (Lys, Gln) with hydrophobic ones (Val, Met), the site becomes more hydrophobic. Alteration of multiple residues gives an engineered LDH that accommodates five or six carbon analogs of lactate and uses them as substrates. 20 STRUCTURAL SCAFFOLDS Relatively few of the amino acid residues in a protein are actually involved in the active site. Most of the protein provides the 3D platform or scaffold needed to correctly position the active site residues. Quite often the scaffold is much larger than really necessary. For example, the β-galactosidase of Escherichia coli (LacZ protein) has approximately 1000 amino acids, whereas most simple hydrolytic enzymes have only 200 to 300. Presumably it should be possible to redesign a functional βgalactosidase that is only 25% to 30% the size of LacZ protein. From an industrial viewpoint, such a smaller protein would obviously be more efficient. 21 DIRECTED EVOLUTION Directed evolution is a powerful technique to alter the function of an enzyme without the need for exhaustive structural and functional data. Directed evolution can be used to change substrate specificity, either changing the enzyme to recognize a totally different substrate, or making subtle changes where the substrate is slightly different. The main premise of directed evolution is the random mutagenesis of the gene of interest, followed by a selection scheme for the new desired function. Directed evolution screens for new enzyme activities by constructing a library of different enzymes derived from the same original protein. Each protein in the library is slightly different because of random mutagenesis. Random mutants may be generated over the entire length of the gene sequence. Alternatively, certain target amino acids can be replaced by random amino acids. The third main method for generating mutants relies on recombination (homologous or nonhomologous). These mutant genes are then screened for the new, desired enzyme activity after insertion into a suitable expression vector and host cell. 22 Random mutagenesis usually starts with a gene whose function is dose to that desired. The gene is randomly mutated throughout the entire sequence using error-prone PCR (epPCR). Different methods exist to induce errors during PCR amplification. The most straightforward is to add MnC1, to the PCR reaction. Taq polymerase has a fairly high rate of incorporating the wrong nucleotide, and MnCI, stabilizes the mismatched bases. The error will be copied in subsequent rounds of amplification. Adding nucleotide analogs such as 8-oxo-dGTP and dITP, which form mismatches on the opposite strand, can also enhance the PCR error rate. These analogs in combination with MnCl2, can induce a wide variety of different mutations along the length of a gene. Some random mutations that occur outside the active site may cause subtle changes with profound effects on substrate recognition and enzyme function. The third method to form. new enzymes via directed evolution involves various schemes for recombining different domains. These are based on homologous or nonhomologous sequences and encompass a variety of different protocols, including DNA shuffling and combinatorial protein libraries. 23 MnCl2 High conc. MgCl2 AGTC Nucleotide Analog mutagen 24 ADDING MEW FUNCTIONAL GROUPS USING NONNATURAL AMINO ACIDS Many nonnatural amino adds have different functional groups that are useful in protein engineering. For example, adding p-benzoyl-L-phenylalanine( pBpa) into one position of glutathione-S-transferase (GST) adds a crosslinking group that can be activated by UV irradiation. When GST is modified in this way, UV irradiation creates a covalently linked homodimer (Fig. 11.6). 25 FIGURE 11.6 Adding New Functional Groups to Proteins (A) Nonnatural amino acids add new functional groups. These can be incorporated into a protein during translation. (B) The nonnatural amino acid, pBpa, crosslinks the GST mutant protein to form a homodimer. 26 Incorporating a nonnatural amino acid into a protein can be done on a small scale by isolating the tRNA for a particular amino acid and attaching the nonnatural amino acid. The charged tRNA is then added to an in vitro protein translation system, which incorporates the nonnatural amino acid into the growing polypeptide chain. The chemical method is too costly and time consuming for large-scale use. For large-scale incorporation, E. coli may be modified to insert the nonnatural amino add in vivo. Inserting a nonnatural amino acid during in vivo protein synthesis requires mutant aminoacyl-tRNA synthetase a that charges a tRNA with the nonnatural amino add. The laboratory of Peter G. Schultz, at the Scripps Research Institute, has developed an E. coli strain that incorporates pBpa at a specific amber codon (One of the three terminator codons. Its sequence is UAG ). Directed evolution was used to mutate a tyrosyl-tRNA synthetase from M. jannaschii. The enzyme from M. jannaschii was used because it does not recognize any endogenous E. coli tRNA. Consequently, it needs the gene for its specific partner tRNA to be provided as well. In addition, the partner tRNA was altered so that it recognizes the amber stop codon instead of its original natural codon (i.e., it is an amber mutant tRNA). The result is that when the mutant tyrosyl-tRNA synthetase is expressed in27 E. coli, it inserts whichever amino acid it is charged with at amber stop codons. 28 To alter the tyrosyl-tRNA synthetase, a library of mutant enzymes was generated by random mutation of each amino acid residue involved in recognition of the amino acid substrate (originally tyrosine). The library of mutant tRNA synthetase genes was transformed into E. coli that possess a gene for the partner tRNA, and a gene for chloramphenicol resistance with an amber codon in the middle. The E. coli were grown in the presence of pBpa and chloramphenicol. If the mutant tRNA synthetase was able to insert an amino acid at the amber stop codon, then the chloramphenicol resistance gene (CAT) was expressed, and the cells lived. Otherwise, the cells died. This was the positive selection (Fig. 11.7). This positive selection does not exclude mutant tRNA synthetases that charge the amber tRNA with a natural amino acid, so a negative selection scheme was used next. The plasmids carrying the mutant tRNA synthetases were isolated and transformed into a different E, coli strain. This toxin gene with an amber suppressor mutation plus the amber tRNA. Here the mutants were grown without E. coli had a any pBpa. If the mutant tRNA synthetase could charge the amber tRNA with a natural amino add, the toxin would be made and the E. coli would die. This eliminated mutant tRNA synthetases that used natural amino adds. The selection scheme was repeated numerous times, and finally a specific mutant tRNA synthetase was isolated (Fig. 11.7) that recognized the amber tRNA 29 and linked pBpa to it. p-benzoyl-L-phenylalanine Positive and Negative Selections for Mutant tRNA Synthetase toxin gene chloramphenicol resistance gene (CAT) 30 FIGURE 11.7 Positive and Negative Selections for Mutant tRNA Synthetase Tyrosyl-tRNA synthetase normally attaches tyrosine to the tRNA for the CUA amber codon. The amino acids that recognize tyrosine were randomly mutagenized to form a library of different tRNA synthetases that still recognize the same tRNA, but might attach different amino acids. Next, these library clones (MutTyrRS) were expressed in a cell containing another plasmid carrying genes for the amber tRNA (Mj tRNACUA) and for chloramphenicol acetyl transferase (CAT). In the middle of the CAT gene is an amber codon (amberCAT). These E. coli are grown with pBpa and chloramphenicol. MutTyrRS must charge the Mj tRNACUA with pBpa or another amino acid to express CAT, which protects the E. coli from chloramphenicol. The library clones that survive this selection are expressed in a different E. coli host (left). This strain has the gene for the amber tRNA (Mj tRNACUA) plus a toxin gene with an amber codon. The toxin protein is made if the MutTyrRS charges the amber tRNA with any amino acid (no pBpa is present). This eliminates clones that charge the amber tRNA with an endogenous amino acid. The positive and negative selection schemes are alternated to find the best mutant tRNA synthetase. 31 RECOMBINlNG DOMAINS A related approach to directed evolution is to deliberately recombine functional domains from different proteins. An example is the creation of novel restriction enzymes by linking the cleavage domain from the restriction enzyme FokI with different sequencespecific DNA binding domains. enzyme FokI is a type II restriction with distinct N-terminal and C-terminal domains that function in DNA recognition and DNA cutting, respectively. By itself the endonudease domain cuts DNA nonspecifically. However, when the nuclease domain is attached to a DNA-binding domain, this domain determines the sequence specificity of the hybrid protein. The two domains may be joined via a sequence encoding a linker peptide such as (GlyGlyGlyGlySer), (Fig. 11.8). Cleavage of the DNA occurs several bases to the side of the recognition sequence, as in the native FoKI restriction enzyme. 32 FIGURE 11.8 Recombining Domains to Create a Novel Endonuclease The FokI endonuclease has separate nuclease and sequence recognition domains. Using genetic engineering, the recognition domain of FokI can be replaced with a Gal4 recognition domain, which binds to a different DNA sequence. The two domains are joined with an artificial linker peptide. The new hybrid enzyme now cuts DNA at different locations from the original FokI protein. 33 Several different DNA binding domains have been combined with the nuclease domain of FokI. For example, the Gal4 protein of yeast is a transcriptional activator that recognizes a 17 base-pain consensus sequence. Gal4 has two domains, a DNA binding domain and a transcription activating domain. The N-terminal 147 amino acids of Gal4 can be fused to the nuclease domain of FokI, giving a hybrid protein that binds to the Gal4 consensus sequence and cleaves the DNA at that location. Zinc finger domains have also been joined to the nuclease domain of FokI. The zinc finger is a common DNA binding motif found in many regulatory proteins. The zinc finger consists of 25 to 30 amino adds arranged around a Zn ion, which is held in place by binding to conserved cysteines and histidines. Each zinc finger motif binds three base pairs, and a zinc finger domain may possess several motifs. In this example, domains approximately 90 amino adds long and comprising three zinc finger motifs, and which therefore specifically recognize nine base DNA sequences, were connected to the FokI nuclease domain (Fig. 11.9). 34 FIGURE 11.9 Assembly of Zinc Finger Domains (A) The nuclease domain of FokI can be linked to a zinc finger domain containing three zinc finger motifs. Zinc fingers recognize three nucleotides each; therefore, any 9-base-pair recognition sequence can in principle be linked to the nuclease domain. (B) The sequence of the hybrid between the FokI nuclease and the zinc finger domain. The letters represent the amino acid sequence. The amino acids in large letters recognize and bind the DNA sequence. Zinc finger motif: 25 aa genome editing 35 DNA SHUFFLING Natural selection works on new sequences generated both by and recombination. mutation DNA shuffling is a method of artificial evolution that includes the creation of novel mutations as well as recombination. The gene to be improved is cut into random segments around 100 to 300 base pairs long. The segments are then reassembled by using a suitable DNA polymerase with overlapping segments or by using some version of overlap PCR. This recombines segments from different copies of the same gene (Fig. 11.10). Mutations may be introduced in several ways, including the standard mutagenesis procedures. In addition, the DNA segments may be generated using error-prone PCR instead of by using restriction enzymes. Alternatively, mutations may be introduced during the reassembly procedure itself by using a DNA polymerase that has impaired proofreading capability. The result is a large number of copies of the gene, each with several mutations scattered at random throughout its sequence. The final shuffled and mutated gene copies must then be expressed and screened for altered properties of the encoded protein. 36 FIGURE 11.10 DNA Shuffling for a Single Gene Introducing point mutations and shuffling gene segments can generate a better version of a protein. First, many copies of the original gene are generated with random mutations. The genes are then cut into random segments. Last, the fragments are reassembled using overlap PCR. The new constructs37 must be assessed for enhanced protein function. FIGURE 11.11 DNA Shuffling for Multiple Related Genes Shuffling segments from related genes can also enhance the function of a particular protein. The original set of related genes are digested into small fragments and reassembled using PCR. The new combinations are tested for a change in function. 38 A more powerful variant of DNA shuffling is to start with several closely related (i.e, homologous) versions of the same gene from different organisms. The genes are cut at random with appropriate restriction enzymes and the segments mixed before reassembly. The result is a mixture of genes that have recombined different segments from different original genes (Fig. 11.11). Note that the reassembled segments keep their original natural order. For example, several related β-lactamases from different enteric bacteria have been shuffled. The shuffled genes were cloned onto a plasmid vector and transformed into host bacteria. The bacteria were then screened for resistance to selected β-lactam antibiotics. This approach yielded improved β-lactamases that degraded certain penidins and cephalosporins more rapidly and so made their host cells up to 500-fold more resistant to these p-lactam antibiotics. 39 COMBINATORIAL PROTEIN LIBRARIES Another approach to protein engineering is to generate large numbers of different protein sequences and then screen them for some useful enzyme activity or other chemical property. (Screening is often done by phage display Antibody: light-chain library and heavy-chain library). Rather than merely generating large numbers of random polypeptides, combinatorial screening usually uses pre-made modules of some sort to create a random shuffling library. For example, protein motifs known to provide a binding site for metal ions or metabolites might be combined with segments known to form structures such as an alpha helix. In a common approach, DNA modules of around 75 base pairs (i.e., 25 codons) are made by chemical DNA synthesis. Several modules are then assembled to give a new artificial gene. The modules are usually joined by PCR using overlapping primers (Chapter 4). Modules may be joined in a chosen order or in a randomized manner. For proper expression of the assembled sequence, the front and rear modules are normally specified to provide suitable promoter and terminator sequences. The intervening modules may then be randomly Generation of shuffled to generate more possible variation (Fig. 11.12). 40 FIGURE 11.12 Generation of Random Shuffling Library To create a library of new proteins, different modules can be randomly joined together. The first module (yellow) has sequences for the promoter; therefore, this is always added at the front. Similarly, the last module (purple) has the terminator sequences. Using overlapping PCR primers, the modules are joined together in a particular order (part A), or randomly (part B). Because random assembly creates many different combinations, this method creates a library of new proteins that can be screened for a particular function or set of functions. 41 Combinatorial library: exon shuffling 42 FIGURE 11.13 Generation of Alternative Splicing Library The exons from an original gene can be recombined such that one exon is missing in each novel construct. The new genes are then screened for new or altered function. 43 ENGlNEERED BINDING PROTEINS Making drugs specific for a particular organ can eliminate many unwanted side effects. One, way of achieving this is to attach the drug to a reagent that recognizes proteins specific to particular tissues. antibodies are the most widely used reagents for binding specific target proteins. However, antibodies require disulfide crosslinks to function, and these are often hard to maintain during large-scale manufacture. Some researchers have therefore been seeking alteratives to antibodies. To generate novel binding domains, a binding protein with a known structure is chosen and the amino acid residues associated with binding are identified. The binding protein is modified by mutation of these residues and then screened for new binding partners. It is hoped that the targeted directed evolution approach will find new, more easily isolated proteins for targeting drugs to specific target cells within our bodies. 44 FIGURE 11.14 Structural Domains Involved in Protein-Protein Binding Some types of protein backbones used as scaffolds for protein-binding agents. The proteins used (with their Protein Data Bank ID numbers) are beta sandwich (1FNA, fibronectin); beta barrel (A chain of 1BBP, lipocalin); thee-helix bundle (1Q2N, SpA domain); repeat proteins (1MJ0, AR protein); peptide binders (chain A of 1KWA, PDZ domain); small scaffolds (chain F of 1MEY, zinc-finger protein); scaffolds presenting constrained peptides (chain A of 2TrX, thioredoxin A); proteins with intrinsic fluorescence (chain A of 1GFL, GFP) or intrinsic enzyme activity (1M40, beta-lactamase); protease inhibitors (1ECY, ecotin); and disulfide-bonded scaffolds (chain A of 1CMR, scorpion toxin). Cysteine residues and disulfide bonds are depicted in yellow. From: Binz and Plückthun (2005). Engineered proteins as specific binding reagents. Curr Opin Biotechnol16, 459–469. Reprinted with permission. 45 Monoclonal Antibodies as Therapeutic Agents About 100 years ago, horses were inoculated with the bacterium Corynebacterium diphtherial However, the administration of horse antiserum containing antibodies against the exotoxin provides passive immunity, protecting the patient from a fatal outcome when the antiserum is given within the first few days of the onset of infection. Patients often develop their own antibodies against the foreign proteins of either whole or partially purified therapeutic antiserum. After a second treatment, the sensitized patient may go into anaphylactic shock and die. Today, with the advent of hybridoma methodology (mouse antibodies), antibodies are once again seen as potential therapeutic agents. This technique can be used to maintain a continuous supply of pure monospecific antibody. Therefore, the present goal is to design and produce human monoclonal antibodies with both specific immunotherapeutic properties and lowered potential for immunogenicity. Structure and Function of Antibodies An antibody molecule (immunoglobulin) consists of two identical "light" (L) protein chains and two identical "heavy" (H) protein chains. The N-terminal regions of the L and H chains together form the antigen recognition site of each antibody. (Fig. 10.5). The sites that recognize and bind antigens consist of three complementarity-determining Figure 10.5 Structure of an regions (CDRs) that lie within the antibody molecule. The H and L variable (VH and VL) regions at the chains contain variable (VL and VH) N-terminal ends of the two H and two and constant (CL, CH1, CH2, and CH3) L chains. The CDRs are the part of domains with their CDRs (CDR1, an antibody molecule with the CDR2, and CDR3). The Fv, Fab, and greatest variability in amino acid Fc portions of an antibody molecule are delineated. The N-terminal (NH2) sequence. and C-terminal (COOH) ends of each polypeptide chain are indicated. Fc portion elicits several immunological responses after antigen-antibody binding occurs: The complement cascade is activated. The components of this system break down cell membranes, activate phagocytes, and generate signals to mobilize other components of the immunological response system. Antibody-dependent cell-mediated cytotoxicity (ADCC), which is the result of the binding of the Fc portion of the antibody to an Fc receptor of an ADCC effector cell (natural killer cells, neutrophils, macrophages), is produced. The bound effector cell releases substances that lyse the foreign cell to which the Fab portion of the antibody molecule is bound. After the Fab region binds to a soluble antigen, the Fc portion of an antibody can be bound to Fc receptors of phagocytic cells, which engulf and destroy the antibody-antigen complex. Treating Brain Tumors Anti-EGF receptor Ab Human Monoclonal Antibodies Although the initial studies of immunotherapeutic agents were promising, there are drawbacks to chemical coupling and the use of a nonhuman monoclonal antibody. Finally, if the therapy requires multiple treatments, the antibody component should be from a human source to prevent immunological cross-reactivity and sensitization of the patient. It is very difficult to create specific non-cross-reactive antibodies because of problems associated with obtaining human monoclonal antibodies by conventional hybridoma techniques. 1. The human chromosomes of fused human lymphocyte-mouse myeloma cells are unstable, and so cells producing a human monoclonal antibody are extremely rare. 2. No effective human myeloma cell line that can replace the mouse myeloma cell line in the procedure has been discovered. 3. For ethical reasons, it is not acceptable to immunize humans against various antigens. In one scheme, human B lymphocytes that were actively producing antibody were isolated from subjects and treated with a fluorescencelabeled antigen. Fluorescence-activated cell sorting (FACS) was then used to enrich the cell sample for specific antibody-producing B lymphocytes. Because B cells do not grow well in culture, they were transformed with Epstein-Barr virus, which allows them to grow more readily in culture. Some of these transformed B-cell clones were found to secrete human monoclonal antibody that interacted with the selecting antigen. Unfortunately, with this strategy, the yields tend to be small and the monoclonal antibodies have weak antigen binding affinities. In addition, there is a low probability that a nonimmunized individual will have antibodysecreting cells that recognize the selecting antigen. It may also be possible to introduce cells of the human immune system into a mutant mouse strain that lacks, for the most part, its own natural immunological cell repertoire. After the transplantation of human immune system stem cells, such a mouse, with severe combined immunodeficiency (scid mouse), acquires a human cell immune system and, in response to challenge by antigen, can produce human antibodies. Other workers are attempting to introduce human immunoglobulin genes into the germ line of mice to create transgenic mice that could produce a human immunoglobulin after immunization with a particular antigen. Standard hybridoma methodology could be used to isolate cells that secreted a specific monoclonal antibody from the transgenic animals; then these positive cell lines could be screened to determine which lines produced antibody that was encoded by the human immunoglobulin transgenes. In fact, a transgenic mouse that expresses the native form of both H and L human immunoglobulin genes has recently been reported. The humanization of the immune system of scid mice and the generation of transgenic mouse lines are both laborious ways to produce human monoclonal antibodies. Consequently, researchers have been examining genetic engineering strategies to create both human therapeutic antibodies and effective dual-function proteins that have the ability to bind a target and then destroy it. Hybrid Human-Mouse Monoclonal Antibodies The modular nature of antibody functions has made it possible to convert a mouse monoclonal antibody into one that has some human segments but still retains its original antigen binding specificity. This hybrid antibody is called a chimeric antibody, or a "humanized" antibody. The first portion of a mouse monoclonal antibody that was targeted for replacement with a human sequence was the mouse Fc fragment. It was chosen because the mouse Fc fragment functions poorly as an effector of immunological responses in humans; it is also the most likely fragment to elicit the production of human antibodies. When a chimeric antibody that contained the binding site from a mouse monoclonal antibody directed against the surface of human colon cancer cells was tested in patients with colorectal cancer, it remained in the blood system about six times longer than the complete mouse monoclonal antibody did, thereby extending the period of effectiveness. Only 1 patient of the 10 developed a mild antibody response to the chimeric antibody. To diminish immunogenicity and to introduce human Fc effector capabilities, the DNA coding sequences for the Fv regions of both the L and the H chains of a human immunoglobulin were substituted for the Fv DNA sequences for the L and H chains from a specific mouse monoclonal antibody (Fig. 10.9). This replacement of Fv coding regions can be accomplished either by using oligonucleotides and in vitro DNA replication or by using subcloned segments. The DNA constructs for both chimeric chains were cloned into an expression vector and transfected into cultured B lymphocytes (????) from which the chimeric antibody was collected. The "humanizing" of mouse and rat monoclonal antibodies has been taken one step further than the formation of chimeric molecules described above by substituting into human antibodies only the CDRs of the rodent monoclonal antibodies (Fig. 10.10). Because these "reshaped" human antibodies have antigen binding affinities similar to those of the original rodent monoclonal antibodies, they may be effective therapeutic agents. Figure 10.10 Genetically engineered humanized antibody. The CDRS (CDR1, CDR2, and CDR3) from the genes for H and L immunoglobulin chains of a mouse monoclonal antibody replace the CDRS of the genes for a human antibody. The product of this constructed gene is an immunoglobulin with the antigen binding specificity of the mouse monoclonal antibody (pink) and all the other properties of a human antibody molecule (light blue). Humanizing rodent monoclonal antibodies may be performed as follows. Starting with a rodent hybridoma cell line, cDNAs for the L and H chains can be isolated. degenerate primers (VL and VH) signal sequences constant regions The variable regions of these cDNAs can be amplified by PCR. The oligonucleotide primers that are used for this amplification are complementary to the sequences at the 5' and 3' ends of the DNA encoding the variable regions, where there is a high degree of nucleotide sequence conservation from one antibody gene to another. From the nucleotide sequences of the cDNAs for the light and heavy regions (VL and VH), it is possible to delineate the limits of the CDRs. It is usually straightforward to determine where the CDRs begin and end, since these regions are highly variable in sequence while the sequences of the framework regions are relatively conserved.