Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Catalytic triad wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Expression vector wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Magnesium transporter wikipedia , lookup
Interactome wikipedia , lookup
Protein purification wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Point mutation wikipedia , lookup
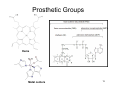
Ancestral sequence reconstruction wikipedia , lookup
Western blot wikipedia , lookup
Genetic code wikipedia , lookup
Biosynthesis wikipedia , lookup
Metalloprotein wikipedia , lookup
Peptide synthesis wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Homology modeling wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Two-hybrid screening wikipedia , lookup
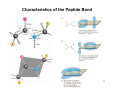
MCAT Question • Covalent bonds are the strongest chemical bonds contributing to the protein structure A peptide bond is formed between with of the following? A. B. C. D. Carboxylic group and amino group Two carboxylic groups Two amino groups Ester group and ammonium group 1 But some amino acids have multiple properties • Y • K OH? Hydrogen bond donor or acceptor 2 Quiz 3 The Peptide Bond • is usually found in the trans conformation • has partial (40%) double bond character • is about 0.133 nm long shorter than a typical single bond but longer than a double bond • Due to the double bond character, the six atoms of the peptide bond group are always planar! 4 Characteristics of the Peptide Bond 5 Proteins - Large and Small • Insulin - A chain of 21 residues, B chain of 30 residues -total mol. wt. of 5,733 • Glutamine synthetase 12 subunits of 468 residues each - total mol. wt. of 600,000 • Connectin proteins alpha - MW 2.8 million! • beta connectin - MW of 2.1 million, with a length of 1000 nm -it can stretch to 3000 nm! 6 The sequence of ribonuclease A 7 The Sequence of Amino Acids in a Protein • is a unique characteristic of every protein • is encoded by the nucleotide sequence of DNA • is read from the amino terminus to the carboxyl terminus 8 Architecture of Proteins • Shape - globular or fibrous • The levels of protein structure - Primary - sequence - Secondary - local structures - H-bonds - Tertiary - overall 3-dimensional shape - Quaternary - subunit organization 9 “Protein” One or more polypeptide chains • One polypeptide chain - a monomeric protein • • • • • More than one - multimeric protein Homomultimer - one kind of chain Heteromultimer - two or more different chains Hemoglobin, for example, is a heterotetramer It has two alpha chains and two beta chains 10 What forces determine the structure? • Primary structure - determined by covalent bonds • Secondary, Tertiary, Quaternary structures determined by weak forces and disulfide bonds • Weak forces - H-bonds, ionic interactions, van der Waals interactions, hydrophobic interactions 11 Other Chemical Groups in Proteins Proteins may be "conjugated" with other chemical groups • If the non-amino acid part of the protein is important to its function, it is called a prosthetic group. • glycoprotein, lipoprotein, nucleoprotein, phosphoprotein, metalloprotein, hemoprotein, flavoprotein. 12 Prosthetic Groups Heme Metal centers 13 Sequence Determination Frederick Sanger was the first - in 1953, he sequenced the two chains of insulin. • Sanger's results established that all of the molecules of a given protein have the same sequence. • Proteins can be sequenced in two ways: - amino acid sequencing - sequencing the corresponding DNA in the gene The sequence shown is that of bovine insulin. 14 Determining the Sequence An Eight Step Strategy • • • • • 1. Cleave (reduce) disulfide bridges 2. If more than one polypeptide chain, separate. 3. Determine composition of each chain 4. Determine N- and C-terminal residues 5. Cleave each chain into smaller fragments and determine the sequence of each chain • 6. Repeat step 5, using a different cleavage procedure to generate a different set of fragments. • 7. Reconstruct the sequence of the protein from the sequences of overlapping fragments • 8. Determine the positions of the disulfide crosslinks 15 Step 1: Cleavage of Disulfide bridges • Performic acid oxidation • Sulfhydryl reducing agents - mercaptoethanol - dithiothreitol or dithioerythritol - to prevent recombination, follow with an alkylating agent like iodoacetate 16 Step 2: Separation of chains • Subunit interactions depend on weak forces • Separation is achieved with: - extreme pH - 8M urea - 6M guanidine HCl - high salt concentration (usually ammonium sulfate) 17 Step 3: Determine Amino Acid Composition • The complex amino acid mixture in the hydrolysate obtained after digestion of a protein in 6 N HCl can be separated into the component amino acids by either ion exchange chromatography (separation by charge) or reverse-phase chromatography (separation by polarity) • Both of these methods of separation and analysis are fully automated in instruments called amino acid analyzers. Analysis of the amino acid composition of a 30-kD protein by these methods requires less than 1 hour and only 6 mg (0.2 nmol) of the protein. • results often yield ideas for fragmentation of the 18 polypeptide chains (Step 5, 6) Problems with Acid hydrolysis aa composition quantification • S and T degrade, but the data from different time points can be extrapolated to determine the composition (Fig. 5.12) • Asn and Gln are converted to Asp and Glu, respectively, because the amide linkages are acid labile – results are usually reported for Asx and Glx 19 Step 4: • Identify N- and C-terminal residues N-terminal analysis: – Edman's reagent – phenylisothiocyanate – derivatives are phenylthiohydantions – or PTH derivatives Efficiency of reaction cycles between 10-50 (an amino acid each cycle) depending on protein properties (e.g molecular weight) 20 Step 4: Identify N- and C-terminal residues • C-terminal analysis – Enzymatic analysis (carboxypeptidase) – Carboxypeptidase A cleaves any residue except Pro, Arg, and Lys – Carboxypeptidase B (hog pancreas) only works on Arg and Lys – Carboxypeptidase C and Y cleave any residue – Exopeptidases cleave from the termini 21 Steps 5 and 6: Fragmentation of the chains • Enzymatic fragmentation – – – – Trypsin - cleavage on the C-terminal side of Lys, Arg Chymotrypsin - C-terminal side of Phe, Tyr, Trp Clostripain - like trypsin, but attacks Arg more than Lys Staphylococcal protease • C-terminal side of Glu, Asp in phosphate buffer • specific for Glu in acetate or bicarbonate buffer • Endopeptidases cleave within the protein sequence – some are non-specific such as pepsin and papain • Chemical fragmentation - cyanogen bromide (CNBr) – acts only on methionine residues – is useful because proteins usually have only a few Met residues 22 Enzymatic Cleavage e.g. Trypsin 23 Mechanism of CNBr 24 25 Step 7: Reconstructing the Sequence • Use two or more fragmentation agents in separate fragmentation experiments • Sequence all the peptides produced (usually by Edman degradation) • Compare and align overlapping peptide sequences to learn the sequence of the original polypeptide chain 26 Reconstructing the Sequence Compare cleavage by trypsin and staphylococcal protease on a hypothetical peptide: • Trypsin cleavage: A-E-F-S-G-I-T-P-K L-V-G-K • Staphylococcal protease: F-S-G-I-T-P-K L-V-G-K-A-E 27 Reconstructing the Sequence • The correct overlap of fragments: L-V-G-K A-E-F-S-G-I-T-P-K L-V-G-K-A-E F-S-G-I-T-P-K • Correct sequence: L-V-G-K-A-E-F-S-G-I-T-P-K 28 Sequence analysis of catrocollastatin-C, a 23.6 kD protein from the venom of Crotalus atrox 29 MCAT Question • Pepsin, trypsin, and chymotrypsin cleave polypeptides into fragments at a specific point in the middle of the chain. These enzymes are properly characterized as: A. B. C. D. endopeptidases zymogens ligases exopeptidases 30 Nature of Protein Sequences • Sequences and composition reflect the function of the protein • Membrane proteins have more hydrophobic residues, whereas fibrous proteins may have atypical sequences 31 Frequencies of amino acids in proteins Legend: gray = aliphatic, red = acidic, green = small hydroxy, blue = basic, black = aromatic, white = amide, yellow = sulfur 32 Number of aa differences among cytochrome c sequences Homologous proteins from different organisms have similar sequences 33 Phylogeny of Cytochrome c • The number of amino acid differences between two cytochrome c sequences is proportional to the phylogenetic difference between the species from which they are derived • This observation can be used to build phylogenetic trees of proteins • This is the basis for studies of molecular evolution 34 The Role of the Sequence in Protein Structure All of the information necessary for folding the peptide chain into its "native” structure is contained in the primary amino acid structure of the peptide. Interactions between amino acids and backbone atoms stabilize protein structure 35 The Weak Forces What are they? What are the relevant numbers? • van der Waals: 0.4 - 4 kJ/mol • hydrogen bonds: 12-30 kJ/mol • ionic bonds: 20 kJ/mol • hydrophobic interactions: <40 kJ/mol 36 How do proteins recognize and interpret the folding information? • Certain loci along the chain may act as nucleation points • Protein chain must avoid local energy minima • Chaperones may help 37 Consequences of the Amide Plane Two degrees of freedom per residue for the peptide chain • Angle about the C(alpha)-N bond is denoted phi Φ • Angle about the C(alpha)-C bond is denoted psi Ψ • The entire path of the peptide backbone is known if all phi and psi angles are specified • Some values of phi and psi are more likely than others. 38 Steric Constraints on phi & psi Unfavorable orbital overlap precludes some combinations of phi and psi • phi = 0, psi = 180 is unfavorable • phi = 180, psi = 0 is unfavorable • phi = 0, psi = 0 is unfavorable 39 Steric Constraints on phi & psi • G. N. Ramachandran was the first to demonstrate the convenience of plotting phi,psi combinations from known protein structures • The sterically favorable combinations are the basis for preferred secondary structures 40 Classes of Secondary Structure • • • • • All these are local structures that are stabilized by hydrogen bonds Alpha helix Other helices Beta sheet (composed of "beta strands") Tight turns (aka beta turns or beta bends) Beta bulge 41 The Alpha Helix • First proposed by Linus Pauling and Robert Corey in 1951 • Identified in keratin by Max Perutz • A ubiquitous component of proteins • Stabilized by H-bonds 42 The Alpha Helix • Residues per turn: 3.6 • Rise per residue: 1.5 Angstroms • Rise per turn (pitch): 3.6 x 1.5A = 5.4 Angstroms • phi = -60 degrees • psi = -45 degrees 43 The Beta-Pleated Sheet Composed of beta strands • • • Also first postulated by Pauling and Corey, 1951 Strands may be parallel or antiparallel Rise per residue: – 3.47 Angstroms for antiparallel strands – 3.25 Angstroms for parallel strands – Periodicity of two residues 44 The Beta Turn • • • (aka beta bend, tight turn) allows the peptide chain to reverse direction carbonyl C of one residue is H-bonded to the amide proton of a residue three residues away proline and glycine are prevalent in beta turns 45 The β-bulge 46 Amino acids have secondary structure propensities Chou – Fasman Helix and Sheet Propensities (Pα and Pβ) of the Amino Acids Amino Acid Pα Pβ A Ala 1.42 0.83 C Cys 0.70 1.19 D Asp 1.01 0.54 E Glu 1.51 0.37 F Phe 1.130 1.38 G Gly 0.57 0.75 H His 1.00 0.87 I Ile 1.08 1.60 K Lys 1.16 0.74 L Leu 1.21 1.30 M Met 1.45 1.05 N Asn 0.67 0.89 P Pro 0.57 0.55 Q Gln 1.11 1.10 R Arg 0.98 0.93 S Ser 0.77 0.75 T Thr 0.83 1.19 V Val 1.06 1.70 W Trp 1.08 1.37 Y Tyr 0.69 1.47 Source: Chou, P. Y., and Fasman, G. D., 1978. Annual Review of Biochemistry 47:258. 47