Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
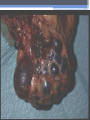
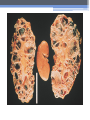
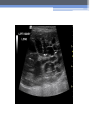
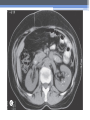
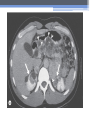
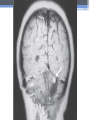
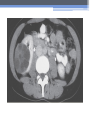
Inherited Kidney Diseases Zehra Eren M.D. Nephrology Department LEARNING OBJECTIVES Recognize • Renal cystic disorders -Autosomal dominant polycystic kidney disease -Autosomal recessive polycystic kidney disease -Medullary sponge kidney -Medullary cystic kidney disease -Tuberous sclerosis cpmplex -von Hippel-Lindau disease LEARNING OBJECTIVES • Alport’s Syndrome • Thin basement membrane disease • Anderson-Fabry disease • Nail-Patella Syndrome • Sicle Cell Nephropathy AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD) • Epidemiology The most common renal hereditary disease, affects 1 in 400 to 1,000 live births Affects all races with equal frequence Autosomal dominant polycystic kidney disease • Prevalence: 1:300 to 1:1000 • 90% of cases are inherited, 10% are sporadic • Only1 to 5% nephrons developed cysts • Cysts are in medulla and cortex • ADPKD causes symptoms in third or fourth decade AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD) • Inheritance - Mutations in the polycystic kidney disease (PKD)1 gene:85%, located on the short arm of chromosome 16 (16p.3.3), codes for a 4,304amino-acid protein (polycystin 1) - Mutations in the PKD2 gene:15%, located on chromosome 4 (4q.21.2), codes for a 968amino-acid protein (polycystin 2) ADPKD • Pathology - massive enlargement of the kidneys secondary to cyst growth and development • Diagnosis -Radiologic Radiological imaging is required (ultrasound is the current imaging modality of choice in at-risk individuals (positive family history in a parent). -Genetic ADPKD • Pathology - massive enlargement of the kidneys secondary to cyst growth and development • Diagnosis -Radiologic Radiological imaging is required (ultrasound is the current imaging modality of choice in at-risk individuals (positive family history in a parent). -Genetic Ravine Criteria for Diagnosing ADPKD Clinical Manifestations • Renal Chronic flank pain Acute pain indicates: infection (pyelonephritis- pyocyst) urinary tract obstruction sudden hemorrhage into cysts Hematuria Impaired renal concentrating ability Nephrolitiasis in 15%to 20% Hypertension in 75% adults Clinical Manifestations • Extrarenal Hepatic cysts: Liver cysts present approximately 10 years after renal cysts Intracranial aneurysms:Occur in 4% to 8% of symptomatic ADPKD patients Cardiac disease:Mitral and aortic prolapse and regurgitation Diverticular disease: Hernias:abdominal and inguinal hernias (up to 45% of ADPKD patients) AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE (ARPKD) • Epidemiology -Affects 1/10,000 to 1/40,000 individuals • Inheritance -Autosomal recessive disorder -Mutations in a single gene on the short arm of chromosome 6p 21.1 • Pathology -The protein encoded by the PKHD1 gene is called polyductin or fibrocystin. Diagnosis and Clinical Manifestations ● Prenatally: Oligohydramnios, enlarged kidneys, and lung hypoplasia with resultant Potter facies ● Infancy: Pneumomediastinum, pneumothorax, HTN, cardiac hypertrophy,endomyocardiofibrotic congestive heart failure, and renal failure ● Older children: Hepatic fibrosis, portal HTN, and complications of variceal bleeding, thrombocytopenia, and anemia predominate TUBEROUS SCLEROSIS COMPLEX (TSC) • Genetics -autosomal dominant pattern -high rate of spontaneous mutations (65% to 75% of patients) • TSC1 →9 (9q34), its protein product is called Hamartin • TSC2→16 (16p13.3),its protein product is named Tuberin bilateral angiomyolipomas MEDULLARY CYSTIC KIDNEY DISEASE (MCKD) • Rare inherited cystic disease, autosomal dominant inheritance: - MCKD1 was mapped to chromosome 1 (1q21) and accounts for the minority of cases - MCKD2 was mapped more recently to chromosome 16 (16p12) and accounts for mutations in most cases MEDULLARY CYSTIC KIDNEY DISEASE (MCKD) • Pathology -normal- to small sized kidneys -cysts located at the corticomedullary junction and in the medulla. However, the presence of cysts is not universal -Microscopically: diffuse tubulointerstitial inflammation, hypertrophied and dilated tubules. Glomeruli are usually normal Diagnosis and Clinical Course • Clinical features with a family history • The presence of cysts supports the diagnosis but is not essential • Computed tomography scan is the most sensitive technique for cyst detection • Polyuria and polydipsia • Progressive renal failure ultimately leads to ESRD von Hippel-Lindau disease • autosomal dominant syndrome manifested by a variety of benign and malignant tumors • VHL gene abnormality is present in about 1 in 36,000 newborns von Hippel-Lindau disease • Clear cell RCCs occur in approximately 70 percent of VHL patients who survive to 60 years of age. • Annual imaging of the kidneys with MRI or CT is indicated to establish the diagnosis • For patients in whom an RCC is diagnosed, we recommend a nephronsparing approach to remove lesions that are 3 cm or larger whenever possible ALPORT’S SYNDROME OR HEREDITARY NEPHRITIS • Genetics -Prevalence of genetic mutation estimated at 1 in 5,000 to 1 in 10,000. -Accounts for 1% to 2% of ESRD cases. -X-linked inheritance in almost all cases (85%) -Of the non–X-linked cases, most are autosomal recessive Alport syndrome • Pathogenesis ALPORT SYNDROME • Renal Manifestations -Hematuria: the characteristic clinical feature of Alport’s syndrome -Proteinuria • Extrarenal Manifestations -Sensorineural hearing loss -Ocular defects: anterior lenticonus -Leiomyomatosis of the esophagus and genitalia THIN BASEMENT MEMBRANE DISEASE (TBMD) • Inherited renal disease of the GBM clinically characterized by persistent microscopic hematuria • inherited in an autosomal dominant fashion, is not accompanied by extrarenal manifestations, and has a benign course • The diagnosis is established by renal biopsy and electron microscopy ANDERSON-FABRY DISEASE (AFD) • X-linked lysosomal storage disease ANDERSON-FABRY DISEASE (AFD) • Diagnosis: measurement of plasma or leukocyte α-GALAactivity, skin biopsy, examination of urine sediment, or sequencing of the defective gene • Clinical Manifestations ▫ Renal ▫ Cardiac: cardiomyopathy Valvular disease Coronary artery disease Nail-Patella syndrom (NPS) osteo-onychodysplasia • Genetics -mutations of the LMX1B gene located at the distal end of the long arm of chromosome 9 -LMX1B is a transcription factor of the LIMhomeodomain type that plays an important role for limb and renal development -incidence:22 per million Nail-Patella syndrom (NPS) • Renal manifestations: • present in one-half of patients - proteinuria (sometimes nephrotic range proteinuria) - Hematuria - Hypertension - Impaired urinary concentration • About 30 percent of patients with renal manifestations will develop end-stage renal disease (ESRD) Sickle Cell Nephropathy • Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of sickle cell disease • The primary event leading to renal involvement appears to be sickling of erythrocytes in the vasa recta capillaries leading to microthrombotic infarction and extravasation of blood in the medulla • Renal failure may ensue in over 10 percent of affected patients, but other clinically significant renal manifestations occur much more frequently Renal Findings in SCN • Hematuria -medullary congestion -renal papillary necrosis -medullary calcification -medullary carcinoma • Tubular Dysfunction -concentration defect -increased sodiım and phosphate reabsorption -decreased proton and potassium secretion -increased urate secretion • Glomerular sclerosis -hyperfiltration -glomerular hypertrophy -proteinuria/nephrotic syndrome -Focal segmental glomerulosclerosis SUGGESTED READING • Goldman's Cecile Medicine, Goldman L, Schafer AI • Case files Internal Medicine, Toy Patlan • Current Medical Diagnosis and Treatment, Maxine A. Papadakis, Stephen J. McPhee, Eds. Michael W. Rabow, Associate Ed. • Current Diagnosis & Treatment: Nephrology & Hypertension S. Berns, Allen R. Nissenson Edgar V. Lerma, Jeffrey