Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Discovery and development of beta-blockers wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Cannabinoid receptor antagonist wikipedia , lookup
Compounding wikipedia , lookup
Plateau principle wikipedia , lookup
Nicotinic agonist wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Psychopharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription costs wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmacognosy wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Drug discovery wikipedia , lookup
Drug design wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Neuropharmacology wikipedia , lookup
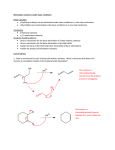
Basic concepts in clinical pharmacology Tony Fox _________________________________ Anthony W Fox MSc MBBS MD(Lond) FRCP FFPM DipPharmMed MCSFS DipFHI CBiol Visiting Professor, Institute of Pharmaceutical Sciences School of Biomedical Sciences, King’s College London. [email protected] Types of drug Pharmacodynamics vs. pharmacokinetics PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG What is ADME ? Lipid membranes Molecular weight, pH, pK, and LogP Liver and kidney Clinical example: The treatment of aspirin overdose PHARMACODYNAMICS: WHAT THE DRUG DOES TO THE BODY Types of drug target Receptors, enzymes, ion channels, and ‘second messengers’ Receptor occupancy – response relationships and theories Agonists, partial agonists Competitive antagonists, uncompetitive antagonists Quantitation of drug effect / antagonism Power, potency and selectivity Unwanted drug effects: concentration-dependent and –independent PK – PD RELATIONSHIPS The classic single-dose time-> concentration curve Chronopharmacology and ‘hysteresis’ Clinical examples : Heroin, morphine, buprenorphine, and codeine : ’Beta blockers’ TYPES OF DRUG Types of drug: or how, basically, do drugs work ? Extension of nutrition: Oxygen, vitamin C, etc. Non-specific Membrane effects: Alcohol, inhaled anaesthetics Enzyme inhibition: ACE inhibitors, physostigmine, sulfonamides Cell surface receptors: Beta blockers, opioids, monoclonals, many others Nuclear receptors and nucleic acids: Steroids, antisense drugs, chemotherapy Ion channel interference: Local anaesthetics, anti-epileptic drugs ‘Chelation’ of endogenous ions / molecules: heparins, tetracyclines PHARMACOKINETICS: BASIC CONCEPTS PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG: VARIOUS ROUTES OF ADMINISTRATION AND FORMULATIONS A Absorption D Distribution M E Metabolism Elimination PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG: Getting from the site of absorption to sites of action, metabolism and elimination A Absorption D Distribution M E Metabolism Elimination PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG: S+E+R A D M E Metabolism ↔ SE + R P + R’ + E Absorption Circulation Distribution Tissue(s) Elimination TYPICAL “Two-compartment” PHARMACOKINETIC MODEL A Typical Time-> Plasma concentration curve Venous plasma sumatriptan concentration (ng/mL) (oral sumatriptan; normal volunteer) Cmax (= 50 ng/mL) 50 40 Absorption , distribution and elimination phase What is the half-time of elimination ? Elimination phase 30 Decay curve ‘1st order elimination’ 20 15 10 7.5 3.75 0 1 Dose (50 mg) 2 3 4 Tmax(= 2 h) 5 6 7 8 9 Time post-dose (hours) 10 11 12 NOTE:- There are exceptions. Some drugs don’t exhibit 1st order elimination (usually at relatively high doses) WHY ? Drug Metabolism: Usually pathways that are used by liver to defend the body against ingested xenobiotics Aim is typically to increase polarity of drug molecule so as to increase urinary and biliary excretion Cytochrome P450 (CYP; ‘sip’) enzymes are especially important, supporting drug metabolism, usually with NADPH, NADH, or flavoprotein coenzymes: Examples De-alkylation: Codeine, diazepam, imipramine Hydroxylation: Phenytoin, ibuprofen, ciclosporine N-oxidation: chlorpheniramine, pethidine, dapsone Sulfation: Paracetamol (acetaminophen), steroids Glucuronidation: morphine, lorazepam Deamination: diazepam, amphetamine Acetylation: isoniazid, sulfonamides Methylation: L-dopa, captopril (Mixed function oxidases) PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG: A D M E ELIMINATION OUTSIDE INSIDE How can drugs get inside that ? A. They don’t: stimulate receptors on the surface B. Actively transported in by a trans-membrane protein C. Passively get in if sufficiently lipophilic LIPOPHILICITY: 1. Electro-negative or –positive substituents can alter the lipophilicity markedly among a series of congeners practolol 2. Non-ionized drugs cross lipid membranes more easily than ionized ones. DRUG CHEMISTRY: GET THE UNITS RIGHT 0. 1 mol = 6.022 x 1023 molecules of a pure substance Avagadro’s constant (L) ~ 6.022 x 1023 mol-1 A molar (1 M) solution has a concentration of 1 mol / L 1. pH: the negative logarithm of the ambient hydrogen ion concentration: H3O+ + OH- <-> 2H2O For pure water [H3O+] is about 0.1 μM => pH = 7 = a chemical definition of acid/base neutrality (physiological neutrality 7.36 – 7.44) The pK is the pH at which half the drug is ionized Two Options for drug solutions Drug+ + OHpH > 7.0 Basic drug pK > 7.0 OR Drug- + pH < 7.0 H+ Acidic drug pK < 7.0 MEASURING LIPOPHILICITY: the Partition Coefficient Drug + cap and shake x 12 h [Drug] in octanol Log P = Log [Drug] in aqueous (Usually octanol) + phosphate buffer pH=7.4 (Or HPLC using a non-polar column and compare unknown to set of knowns validated by the equilibrium shaking method) CLINICAL EXAMPLE: TREATING ASPIRIN OVERDOSE LESS THAN 4 HOURS PRIOR OBJECTIVES: 1. Reduce drug absorption – stomach washout 2. Hasten aspirin excretion Acetylsalicylate m.w. 138, Log P 2.26, pKa 2.97 (Therefore easily filtered by the glomerulus). To encourage urine flow: administer IV fluids Should bicarbonate or ammonium chloride also be given ? Pharmacodynamics: Basic Concepts PHARMACODYNAMICS: WHAT THE DRUG DOES TO THE BODY Types of drug target Receptors, enzymes, ion channels, and ‘second messengers’ Receptor occupancy – response relationships and theories Agonists, partial agonists Competitive antagonists, uncompetitive antagonists Quantitation of drug effect / antagonism Power, potency and selectivity Unwanted drug effects: concentration-dependent and –independent PK – PD RELATIONSHIPS The classic single-dose time-> concentration curve Chronopharmacology and ‘hysteresis’ Clinical examples : Heroin, morphine, buprenorphine, and codeine : ’Beta blockers’ Types of drug target I Enzymes: Useful targets if circulating or form cell surface glycoproteins, e.g., angiotensin converting enzyme (ACE) ’ Ion channels: Usually represent cell surface targets, e.g. sodium channels and local anaesthetics. Types of drug target II: Classical ‘receptors’ (7-transmembrane, G-protein coupled receptors) where conformational change can lead to intracellular ‘second messenger’ formation Examples: Adrenergic receptors: Alpha-1 (vasoconstriction) : antagonists, antihypertensives prazosin, labetalol Beta-1 (cardiac): antagonists protect against angina, long QT syndrome TdP, antihypertensive propranolol et al Beta-2 (bronchodilation): agonists for asthma, salbutamol etc Serotonin receptors: 5HT-1B: agonists triptans, ergots, acute migraine -1F : antagonists, ondansetron, CT emesis Enkephalin receptors: agonists: opioids, analgesia, respiratory depression, g/i stasis, euphoria, dysphoria antagonists: opioid reversal, opioid blockade, naloxone etc Acetylcholine receptors: muscarinic, vagal tone, g/I motility antagonists: oropharyngeal drying (premeds) : sedatives : chlorpheniramine etc The Indirect Relationship between receptor occupancy and drug effect Drugs are conceived as having two properties: i) RECEPTOR AFFINITY: Ability to bind to the receptor (measured as the KD or Kd, i.e., the concentration (mM or μM) needed to achieve 50% receptor occupancy) And ii) RECEPTOR EFFICACY: The ability, once actually bound, to stimulate second messenger generation (Stephenson’s dimensionless constant e). Confusingly, e is also dependent upon available receptor density (see below). Graphs of mass action become semi-log plots For a drug with Kd = 1 μM Kd is a constant, defined as measured in the absence of any other interfering drug / hormone Kd = 1 μ M -7 -6 -6 Log([Radioligand] (M)) -5 Agonism and Antagonism: The difference between binding to a receptor and activating it. EQUILIBRIAL / REVERSIBLE DRUGS [DR] = 1.0 x k(Effect size) [D] + [R] % Maximum (effect or bound) 100 80 BINDING ‘FULL AGONIST’ (e.g. adrenaline, beta-1 adrenoceptor, heart contractility) EFFECT 60 e=1 40 Kd = EC50 = 1 μM 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 Agonism and Antagonism: The difference between binding to a receptor and activating it. EQUILIBRIAL / REVERSIBLE DRUGS [DR] = 0.6 x k(Effect size) [D] + [R] 100 % Maximum (effect or bound) 80 ‘PARTIAL AGONIST’ (e.g. clonidine, alpha-1 adrenoceptor, blood pressure elevation) BINDING EFFECT 60 e = 0.6 40 Kd = EC50 = 1 μM 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 Agonism and Antagonism: The difference between binding to a receptor and activating it. EQUILIBRIAL / REVERSIBLE DRUGS [CA-R] = 0.0 x k(Effect size) [CA] + [R] 100 % Maximum (effect or bound) 80 ‘COMPETITIVE AGONIST’ (e.g. propranolol, beta-1 adrenoceptor, Heart rate) BINDING 60 e = 0.0 40 Kd = 1 μM, no EC50 20 EFFECT 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 Agonism and Antagonism: The difference between binding to a receptor and activating it. EQUILIBRIAL / REVERSIBLE DRUGS [DR] + [CA-R] = 1.0 x k (Effect) + 0.0 x k (Effect) [D] + [CA] + [R] % Maximum (effect or bound) 100 80 No Antagonist ‘FULL AGONIST’ With and without COMPETITIVE AGONIST With Antagonist 60 e=1 40 Kd = EC50 = 1 μM Apparent EC50 > Kd 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 Agonism and Antagonism: The difference between binding to a receptor and activating it. IRREVERSIBLE ANTAGONISM % Maximum (effect or bound) 100 80 60 40 20 IRREVERSIBLE INACTIVATION OF RECEPTOR: Classically: phenoxybenzamine, alpha-1 adrenoceptors, anti-hypertension Others: Monoclonals at various lymphocyte receptors Carbon monoxide Various other poisons No antagonist BINDING EFFECT With antagonist e = 0.6 Kd = EC50 = 1 μM 0 -9 -8 -7 -6 -5 -4 Log [Adrenaline (M)] + phenoxybenzamine Rx Agonism and Antagonism: The difference between binding to a receptor and activating it. EQUILIBRIAL / REVERSIBLE DRUGS [DR] = 10.0 x k(Effect size) % Maximum (effect or bound) [D] + [R] 100 ‘SUPER AGONIST’ Or ‘AGONIST WITH 80 RECEPTOR RESERVE’ (e.g. noradrenaline, 60 beta-1 adrenoceptor, heart rate) EFFECT BINDING 40 20 e>1 EC50 < Kd Kd = 1 μM (> EC50) 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 PK – PD RELATIONSHIPS A Typical Time-> Plasma concentration curve Venous plasma sumatriptan concentration (ng/mL) (oral sumatriptan; normal volunteer) Cmax (= 50 ng/mL) 50 40 Absorption , distribution and elimination phase What is the half-time of elimination ? Elimination phase 30 Decay curve ‘1st order elimination’ 20 15 10 7.5 3.75 0 1 Dose (50 mg) 2 3 4 Tmax(= 2 h) 5 6 7 8 9 Time post-dose (hours) 10 11 12 WHEN DRUG EFFECT IS DIRECTLY RELATED TO DRUG CONCENTRATION (e.g., many antibiotics, isoflurane, remifentanil) Venous plasma drug concentration (ng/mL) Cmax (= 50 ng/mL) 50 PEAK EFFECT 40 30 20 LAG THRESHOLD CONCENTRATION Duration of effect 10 Time of onset of effect No effect 0 1 Dose (50 mg) 2 3 4 Tmax(= 2 h) 5 6 7 8 9 Time post-dose (hours) 10 11 12 THE DYNAMIC SITUATION: % Maximum effect or concentration 100 80 60 40 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 BUT MANY EXCEPTIONS TO THIS SIMPLE SITUATION: - Big distribution phases - Drug sequestration in tissues (especially lipophilic drugs) - Drug interactions - Slow off-phases of drug-receptor equilibria - Tachyphylaxis / sensitization CLINICAL EXAMPLE: Buprenorphine - administer 0.3 mg IV - no detectable drug in circulation after 10 minutes - analgesia onset at about 15 – 30 minutes - duration of effect 6 – 12 hours THE DYNAMIC SITUATION: ‘PHASE PLANE’ or ‘HYSTERESIS LOOP’ 100 % Maximum effect 80 ‘Persistent effect’ or ‘Hangover effect’ or ‘Washout effect’ 60 40 20 Lag 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 THE DYNAMIC SITUATION: BUPRENORPHINE ‘PHASE PLANE’ or ‘HYSTERESIS LOOP’ 100 ‘Persistent effect’ or ‘Hangover effect’ or ‘Washout effect’ % Maximum effect 80 60 40 20 Lag 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 ADVERSE DRUG EFFECTS CAN HAVE THEIR OWN DOSE-RESPONSE CURVES: (“Type A adverse events”) e.g., paracetamol analgesia and hepatotoxicity 100 Wanted Drug Effect % Maximum effect 80 Unwanted drug effect 60 ‘Therapeutic Window’ 40 Compromise Concentration (or dose) 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 CHECK QUESTIONS 1. What does ADME stand for ? (Four words) 2. NAME STRUCTURES marked 3, (5 + 6) 3. Which of ADME does this organ participate in (in general) ? 4. What might be typical units for Km ? 5. Provide the three missing words: Km is the _______________ of ________________ where the reaction rate is half of the ______________ reaction rate. 6. Whose equation is this ? (Two names) 7. If at T1, [S] is vastly greater than Km, Then what is V1 ? 8. If at T1, [S] is vastly greater than Km, then are V1 and V2 (at T2, few minutes later) likely to be the same or different ? 9. Is sodium acetylsalicylate (aspirin) the salt of an acidic or basic drug ? 10. Is the pK of aspirin greater than or less than 7.0 ? 11. Is amphetamine hydrochloride the salt of a basic or acidic drug ? 12. Is the pK of amphetamine hydrochloride greater than or less than 7.0 ? Venous plasma sumatriptan concentration (ng/mL) 13. After a single oral dose of drug, during the time period when the plasma concentration time curve exhibits first-order elimination (Elimination phase), does the half-time of elimination change ? Absorption , distribution and elimination phase Cmax (= 50 ng/mL) 50 40 Elimination phase 30 2 0 10 0 Dose (50 mg) 1 2 3 4 Tmax(= 2 h) 5 6 7 8 9 Time post-dose (hours) 10 11 12 14. For equilibrial drugs, compared with 4, which is A. Non-competitive antagonism, B. Competitive antagonism, C. Partial Agonism, D. Super agonism 15. What is the arrow pointing at ? % Maximum (effect or bound) 100 3 80 BINDING 4 2 60 EFFECT 1 40 20 0 -9 -8 -7 -6 Log [Drug (M)] -5 -4 16. QUESTION- CLINICAL EXAMPLE: 1. You are the OOH drug information scientist / pharmacist / GP / on-call officer for the pharmaceutical company / nurse / etc. 2. The (male) patient has a longstanding history of kidney stones. They are usually small, they usually pass within 48 h, and the patient has been responsibly using 10 mg morphine (full agonist) tablets PRN for a long time. 3. The patient’s partner phones. Usual problem, renal colic, took morphine tablets 6 h and 2 h ago, still in great pain. Some within-date dihydrocodeine (partial agonist) tablets are in the bathroom cabinet, which she was given to use PRN after an out-patient gynaecological procedure 3 weeks ago. Can her partner take additional dose(s) of dihydrocodeine ? Reading Buxton ILO. Pharmacokinetics and pharmacodynamics: The dynamics of drug absorption, distribution, metabolism, and excretion. In: Brunton LL, Lazo JS, Parker KL(Eds) Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw Hill, 2008; 11th edition ISBN 0-07-142280-3, pp.1-39 Reading Buxton ILO. Pharmacokinetics and pharmacodynamics: The dynamics of drug absorption, distribution, metabolism, and excretion. In: Brunton LL, Lazo JS, Parker KL(Eds) Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw Hill, 2008; 11th edition ISBN 0-07-142280-3, pp.1-39