Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Pharmaceutical industry wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription costs wikipedia , lookup
5-HT3 antagonist wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
NMDA receptor wikipedia , lookup
Drug discovery wikipedia , lookup
Theralizumab wikipedia , lookup
Toxicodynamics wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
5-HT2C receptor agonist wikipedia , lookup
Drug interaction wikipedia , lookup
Drug design wikipedia , lookup
Psychopharmacology wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Cannabinoid receptor antagonist wikipedia , lookup
Nicotinic agonist wikipedia , lookup
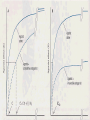
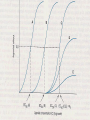
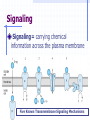
Refresh Your Memory! For which of the following drugs will excretion be most significantly accelerated by acidification of the urine? (a) Weak acid with pKa of 5.5 (b) Weak base with pKa of 3.5 (c) Weak acid with pKa of 7.5 (d) Weak base with pKa of 6.5 DRUG RECEPTORS & PHARMACODYNAMICS Pharmacology-1 Sping semester 2017 Prof.Mayyada Wazaify • Receptor is the component of a cell or organism that interacts with a drug and initiates the chain of biochemical events leading to the drug’s observed effects. • Drug-receptor interactions are studied by pharmacodynamics How receptors allow the understanding of drug actions and clinical uses? 1. Receptors largely determine the quantitative relations between dose or concentration of drug and pharmacologic effects 2. Receptors are responsible for selectivity of drug action 3. Receptors mediate the actions of pharmacologic agonists & antagonists Macromolecular Nature of Drug Receptors: 1. Regulatory proteins – mediate the actions of endogenous chemical signals e.g. neurotransmitters, autacoids, and hormones, as well as many drugs 2. Enzymes – most commonly may be inhibited (e.g., dihydrofolate reductase by methotrexate) 3. Transport proteins (e.g., Na /K + + ATPase inhibited by digitalis) 4. Structural proteins (eg, tubulin, the receptor for colchicine, an anti-inflammatory agent) Relation Between Drug Concentration & Response In vivo: complex In vitro: simple Concentration-Effect Curves & Receptor Binding of Agonists: In vitro, the relation between drug concentration and effect is described by a hyperbolic curve In idealized E max x C or in vitro systems E = ----------------C + EC50 Where E is the effect observed at concentration C; E max is the maximal response that can be produced by the drug, and EC50 is the concentration of drug that produces 50% of maximal effect Concentration-Effect Curves & Receptor Binding of Agonists (continued): = bound Bmax x C B = --------------C + KD The equation describes the relation between drug bound to receptors (B) and the conc. of free drug. Bmax indicates the total conc. of receptor sites (sites bound to the drug at infinitely high conc. of free drug). KD (the equillibrium dissociation constant) is the conc. of free drug at which half-max. binding is observed (reflects the drug affinity) In a reciprocal fashion Graded dose-response curve Graded dose-drug binding efficacy potency affinity Work-out your brain! a. b. c. d. e. A 55-ye old woman with CHF is to be treated with a diuretic drug. Drugs X and Y have the same mechanism of diuretic action. Drug X in a dose of 5 mg produces the same magnitude of diuresis as 500 mg of drug Y. This suggests that: Drug Y is less efficacious than drug X Drug X is about 100 times more potent than drug Y Toxicity of drug X is less than that of drug Y Drug X is a safer drug than drug Y Drug X will have a shorter duration of action than drug Y because less of drug X is present for a given effect Competitive and irreversible antagonists Competitive Antagonists In the presence of a fixed concentration of agonist, increasing concentrations of a competitive antagonist progressively inhibit the agonist response; high antagonist concentrations prevent response completely; Conversely, sufficiently high concentrations of agonist can completely surmount the effect of a given concentration of the antagonist, ie Emax for the agonist remains the same for any fixed conc. of antagonist Competitive Antagonists Schild equation: C’ [I] ----- = 1 + ----C KI Where C’ is the conc. of agonist required to produce a given effect in the presence of a fixed conc. ([I]) of competitive antagonist, C is the agonist concentration required to produce the same effect in the absence of the antagonist; KI is the dissociation constant Irreversible Antagonists The antagonist’s affinity for the receptor is so high that the receptor is unavailable for binding of agonist The number of remaining unoccupied receptors may be too low for the agonist to elicit maximal response If spare receptors are present, however, a lower dose of irreversible antagonist may leave enough receptors unoccupied to allow achievement of maximum response to agonist (with higher agonist conc.) Concentration-Effect Curves & Receptor Binding of Agonists (continued): Plotting the drug effect against the logarithm of the dose or concentration transforms the hyperbolic dose-response curve into a sigmoid curve with linear midportion. This expands scale at low concentrations (where the effect is changing rapidly) and compresses it at high concentration (where the effect is changing slowly) Receptor-Effector Coupling Binding of agonist to the receptor is the first step of pharmacologic response The transduction process between occupancy of receptors and drug response is often termed coupling Occupancy-response coupling efficiency is determined by: - Initial conformational change in the receptor (full agonist versus partial agonist) - Biochemical events that transduce receptor occupancy into cellular response - “spare” receptors Spare Receptors Receptors are said to be “spare” for a given pharmacologic response when the maximal response can be elicited by an agonist at a concentration that does not result in occupancy of the full complement of available receptors Spare receptors are not qualitatively different from nonspare receptors They are not hidden or unavailable, and when occupied, they can be coupled to response Spare Receptors (continued) In practice, to determine the presence of spare receptors, we compare EC50 and Kd. If EC50 is less than Kd, spare receptors are said to exist; This indicates that to achieve 50% of the maximal effect, fewer than 50% of receptors must be activated How could this phenomenon be explained? Spare Receptors Mechanisms: 1. The effect of the drug-receptors interaction may persist for a much longer time than the interaction itself eg, binding of GTP by an intermediate may greatly outlast the agonist-receptor interaction (the “spareness” is temporal) 2. The actual number of receptors may exceed the number of effector molecules available Important Note: The presence of spare receptors increases sensitivity to the agonist because the likelihood of a drug-receptor interaction increases in proportion to the number of receptors available; Spare Receptors (continued) The sensitivity (EC50) of a cell or tissue to a particular conc. of agonist may depend not only on the affinity of the receptor for binding an agonist (KD) but also on the degree of spareness, i.e. the total number of receptors compared to the number actually needed to elicit a maximal biologic response The KD of the agonist-receptor interaction determines what fraction (B/Bmax) of total receptors will be occupied at a given free conc. (C) of agonist regardless of receptor conc. Spare Receptors (cont’d) B ----Bmax C = -------C + KD If we have a cell with four receptors and four effectors, the number of effectors does not limit the maximal response, and the receptors are not spare in number Partial Agonists Partial agonist produces a lower response at full receptor occupancy than do full agonists. Partial agonists produce concentration-effect curves that resemble those observed with full agonists in the presence of irreversible antagonist The failure to produce a “full” maximal response is not due to decreased affinity for binding to receptors How can an agonist be “partial”? The precise mechanism is not known. We can imagine that a receptor can take two shapes: the inactive form, and an active form, in the absence, as well as the presence of bound ligand; A full agonist binds to the active form more tightly than to inactive form; while a partial agonist has an affinity for both active and an inactive forms. As a result, some receptors will remain in inactive form, so that promote less pharmacologic effect N.B. A partial agonist is less efficacious than the full agonist but it could be more, less or of equal potency to the full agonist… How come? Important: Figure 2-4, pp.17 (Katzung, 2004) A problem? Ask Me! Mechanisms of drug antagonism other than at the same receptor 1) Chemical antagonism - one drug binds and inactivates another drug (need not involve a receptor) (eg, protamine sulfate, (+) protein, is an antidote for heparin (-) charged 2) Physiologic antagonism -using opposing regulatory pathways (eg, insulin opposes hyperglycemic effect of glucocorticoids through different receptors). Wake Up! Q: Which of the following statements about spare receptors is MOST correct? a. b. c. d. e. Spare receptors, in absence of a drug, are sequestered in the cytoplasm Spare receptors will be detected if the intracellular effect of drug-receptor interaction lasts longer than the drug receptor interaction itself Spare receptors influence the maximal efficacy of the drugreceptor system Spare receptors activate the effector machinery of the cell without the need for a drug Spare receptors maybe detected by the finding that the EC50 is greater than Kd for the agonist Important notes before you go! Intrinsic activity: the maximum response to an agonist (Emax) Intrinsic activity of a full agonist is always =1 We compare potency of drugs by measuring their EC50 values Partial agonists cannot have spare receptors because they do not produce a maximum response even at full occupancy of a receptor SIGNALING MECHANISMS & DRUG ACTION Signaling Signaling= carrying chemical information across the plasma membrane Five Known Transmembrane Signaling Mechanisms Five basic mechanisms of transmembrane signaling 1) a lipid-soluble ligand crosses membrane and acts on an intracellular receptor 2) a transmembrane receptor located on membrane-spanning enzymes 3) A transmembrane receptor that binds and stimulates a protein tyrosine kinase Five basic mechanisms of transmembrane signaling 4) a ligand-gated transmembrane ion channel that can be induced to open or close by binding of a ligand 5) a transmembrane receptor protein that stimulates a GTP-binding signal transducer protein (G protein), which in turn generates an intracellular second messenger