Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
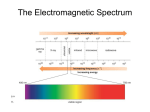
Structure Determination How to determine what compound that you have? One way to determine compound is to get an elemental analysis -basically burn the compound to determine %C, %H, %O, etc. from these percentages can determine the molecular formula Still need to determine structure from molecular formula We have learned various isomers can result from a given molecular formula Consider C3H6O O O H Could have a ketone Different type of carbonyl OH No carbonyl present If we only know the molecular formula, would not know which structure is present Structure Determination Even if a pure sample is obtained, how do we know the actual structure of the compound? The development and improvement of analytical instruments to determine structure has been one of the biggest advancements in organic chemistry during the past 60 years Today almost any structure can be determined with these instruments The important part is to recognize what information each instrument provides, and if deciding between possible isomers which technique can be used to differentiate Techniques to be learned: Mass Spectrometry UV-vis Spectroscopy -mass of compound -conjugation present -isotopes present -distinguish some atoms IR Spectroscopy NMR Spectroscopy -functional groups -bond connectivity of structure -symmetry -most important for structure determination Mass Spectrometry Can determine the molecular weight of a sample and some information about the structure A key part of a mass spectrometry is the need to create a charged species The most common method to create the charged species is electron impact ionization e H H H H C C C H H H H H H H H C C C H H H e H e An electron is accelerated toward a gaseous sample of the compound under consideration Due to the high energy of the electron moving at high speed, an electron is expelled The sample thus is now positively charged, due to the loss of one electron, and is a radical/cation structure Mass Spectrometry The compound (R) thus becomes a radical/cation when bombarded with electrons magnet e R(•+) detector R The radical/cation continues along a path until it reaches a magnetic field Charged species become deflected (are attracted to one magnet) in the presence of the magnetic field and hence the path direction is bent The radius of curvature is dependent upon the mass of the species (m/z), lighter mass species are deflected more and heavier species are deflected less Only a certain mass can thus deflect the correct amount with the curvature of the instrument, heavier species will hit one wall while lighter species will hit the other wall The magnet strength is changed and depending upon when species hit the detector the mass of the compound can be determined Mass Spectrometry The parent ion is called the molecular ion peak (M+) m/z 72 Can find molecular ion, but what are the other peaks? Mass Spectrometry The molecular ion peak can fragment Due to the high energy of the radical/cation generated, this species can fragment CH3CH2 m/z 72 m/z 43 CH3 m/z 57 Remember only the charged species will be detected (the radical species will not be affected by the magnetic field) The probability of obtaining a given fragment is due to the STABILITY of the cations produced Effect of Isotopes Remember that an isotope has the same number of protons and electrons, but a different number of neutrons Since neutrons and protons are the “heavy” parts of an atom, the extra number of neutrons will cause a greater mass In a mass spectrometer we can see the effect of this by peaks above the molecular ion peak (M, M+1, M+2, etc.) The ratio of these peaks is diagnostic for which atoms are present The natural abundance of isotopes is well known M M+1 M+2 H 100% C 98.9% 1.1% S 95% 0.8% Cl 75.5% 24.5% Br 50.5% 49.5% I 100% 4.2% Effect of Isotopes Can distinguish atoms by the ratio of peaks above the molecular ion Especially useful to distinguish which halogen is present Cl Br I m/z 78 M/M+2 = 3 m/z 122 M/M+2 = 1 m/z 170 ~ 3/1 = 1 M/M+2 Mass Spectrometry Nitrogen Nitrogen is also diagnostic in a mass spectrum due to the odd/even parity of the mass Consider small molecules and their corresponding mass CH4 NH3 m/z = 16 m/z = 17 The molecular ion peak for a molecule with one nitrogen is always odd, all other common atoms in an organic compound yield an even mass Fragmentation Behavior of Common Functional Groups Alkenes With an alkene the common fragmentation is to create an allylic carbocation m/z 70 m/z 55 Alcohols Two common effects 1) Loss of water H2 O OH m/z 74 m/z 56 Alcohols 2) α-cleavage OH OH OH CH2CH3 m/z 74 m/z 45 McLafferty Rearrangement Any ketone containing a γ-hydrogen can rearrange to the enol form in a MS O H m/z 100 O H m/z 58 Ketones Ketones can also do α-cleavage similar to alcohols O m/z 100 O O m/z 43 CH2CH2CH2CH3 High Resolution Mass Spectrometry (HRMS) These high sensitivity mass spectrometers, called HRMS, can be used to determine molecular formula a HRMS can detect particle masses with an accuracy of 1/20,000 therefore > 0.0001 amu (atomic mass units) Can use this to distinguish compounds with a similar rough mass but with a different molecular formula 12C 1H 16O 12.0000 amu (by definition) 1.0078 amu 15.9949 amu Many structures may have the same integer value molecular weight, but different molecular formulas For example: O HN NH C4H6O1 C5H10 C3H6N2 70.0418 amu 70.0783 amu 70.0531 amu Structure Determination Using Spectroscopy Need methods to distinguish between possible structures A nondestructive way is to use absorption spectroscopy In a simplified picture: Beam splitter Monochromatic light source sample detector blank The ability of the sample to absorb incident radiation is measured by the difference in absorbance at the detector versus the blank Electromagnetic Spectrum All light travels at a constant speed The difference is the wavelength of the light (which also determines the energy of the light) E = hν = (hc) / λ NMR IR UV-vis Infrared Region Wavelength of infrared radiation is ~800 cm-1 to 4000 cm-1 wavenumbers (wavenumbers correspond to number of wavelengths of light in 1 cm) -common descriptor for IR frequencies by organic chemists As the wavenumber becomes larger the energy increases The energy level of infrared light corresponds to the energy required to cause molecular vibrations Depending upon what type of bond is present determines the exact energy required to cause the vibration The energy of light absorbed therefore indicates what functional group is present Bond Vibration The energy of the infrared light can interact with the resonant vibrational frequency of the bond Since different bonds have different energies, they require different energy to cause vibration consider acetone O O H3C CH3 The carbonyl has a strong dipole H3C CH3 E When electric field aligns with dipole, bond shortens The absorption of the infrared light thus changes the dipole for this bond as it vibrates Infrared Spectroscopy Active versus Inactive IR only causes a vibration if there is a change in dipole during vibration Therefore symmetric bonds are inactive CH3-CH3 the carbon-carbon bond of ethane will not observe an IR stretch Or any other symmetric bond An IR “active” bond is therefore a bond that changes dipole during vibration, While an IR “inactive” bond is a symmetric bond that doesn’t change dipole during vibration Number of Vibrations The number of possible vibrations for a given molecule is determined by the number of atoms present For nonlinear molecules obtain 3N-6 vibrations (N equals number of atoms present) 3N-5 vibrations for linear molecule For example consider acetone again (C3H6O1) Acetone has 10 atoms and is nonlinear Therefore expect 3(10)-6 = 24 vibrations The other vibrations are due to different bonds besides the carbonyl stretching, for example the hydrogens Or bending motions Intensity of Absorbance Intensity of light absorbed by a molecule is related to the dipole of the bond The greater the dipole, the greater the absorbance intensity C-O bond stretches are therefore more intense than C-C stretches O C O Realize the intensity of absorbance is not related to the wavenumber The wavenumber is related to the force constant for the bond vibrating (the stiffness of the bond) Infrared Spectroscopy Factors to be considered in an IR spectrum 1) Position of absorbance (wavenumber) Energy required for absorbance 2) Intensity of absorbance Related to the dipole of the bond 3) Shape of absorbance (broad or sharp peaks) Tells information about the type of bond Infrared Spectroscopy Specific functional groups As mentioned specific functional groups have characteristic absorbance frequencies Consider carbon-carbon bonds Wavenumber (cm-1) C C ~1200 cm-1 C C ~1660 cm-1 C C ~2200 cm-1 As the number of bonds increases between two atoms, the stiffness of the bond increases which results in a harder bond to stretch Infrared Spectroscopy Conjugation lowers the stretching frequency (RESONANCE!!!) Wavenumber (cm-1) ~1640-1680 cm-1 ~1620-1640 cm-1 Whenever a functional group becomes more conjugated (adjacent to double bonds for example) the stretching frequency lowers Infrared Spectroscopy C-H bond stretching As the %s character increases in a bond, the bond becomes stiffer (already saw that sp hybridized C-C bonds are stiffer than sp3 hybridized C-C bonds) Same is true for carbon-hydrogen bonds sp3 hybridized sp2 hybridized sp hybridized 2800-3000 cm-1 3000-3100 cm-1 ~3300 cm-1 Key point: only sp3 hybridized C-H bond stretches are below 3000 cm-1 Infrared Spectroscopy Alcohols and amines Both O-H and N-H bonds are “stiff” bonds Therefore they have a higher vibrational frequencies Alcohol Acid Amine RO-H RCO2-H RN-H ~3300 cm-1 ~3000 cm-1 ~3300 cm-1 In addition, both N-H and O-H bonds are involved in hydrogen bonding therefore each bond will experience a slightly different vibrational frequency Therefore this causes the appearance of a broad peak Infrared Spectroscopy Amine peaks show the same broad features (N-H bonds are also involved in hydrogen bonding) Difference is that often observe a sharp peak in the midst of the broad peak (due to one conformation of hydrogen bonding having a preferential formation) Carbonyl Compounds One of the best diagnostic features of IR is for carbonyl compounds Remember there are many types of carbonyl groups (each can be differentiated only with an IR spectrum) O R R Ketone ν 1700-1730 (cm-1) R O O O H R OH R O NH2 R O OR R Cl Aldehyde Acid Amide Ester Acid chloride 1700-1730 1700-1730 1620-1680 1735-1750 1770-1820 2700-2800 3000 Two peaks Broad peak In addition to the carbonyl stretch, other characteristic peaks can distinguish carbonyl groups that display similar C=O stretching frequencies Carbonyl Compounds Due to the large dipole of carbonyl bonds, all carbonyl groups display strong, relatively sharp peaks C=O large dipole C=C small dipole Most carbonyl stretching frequencies are centered around 1700-1730 cm-1 and can be distinguished easily from alkene stretches (~low 1600’s cm-1) due to both the higher frequency and the more intense absorbance Carbonyl Compounds Some carbonyl stretching frequencies are noticeably different than 1700-1730 cm-1 Esters are one type Esters have an appreciably higher stretching frequency O O ν (cm-1) = 1742 Higher frequency means a “stiffer” bond Carbonyl Compounds What causes a “stiffer” carbonyl bond? Substituents on the carbonyl carbon can affect the C=O bond stretch in two ways: Inductive effect Resonance effect O O R R Y O Y R Y More electronegative Y pulls electron density Lone pair of electrons on Y atom can resonate from carbon, which then pulls electrons from to create a C=Y double bond and thus a C-O oxygen to create a stiffer bond single bond – therefore a weaker C-O bond The question is which effect is larger Generally the greater difference in electronegativity between C and Y causes inductive effect to become dominant Y ν (cm-1) Stronger effect Cl 1810 inductive OR 1735 inductive NH2 1660 resonance Carbonyl Compounds Amide group lowers the frequency due to the resonance effect If a nitrogen is attached to the carbonyl carbon then the lone pair of electrons on nitrogen can stabilize the resonance form O O NH2 NH2 Due to this lower energy resonance form the carbonyl carbon-oxygen bond is less “stiff”, therefore the stretching frequency is LOWER Carbonyl Compounds Resonance with extra conjugation will also lower the stretching frequency for a carbonyl O H ν = 1721 cm-1 O H ν = 1699 cm-1 Resonance allows delocalization of π electrons, therefore carbonyl is less “stiff” O O H H Carbonyl Compounds As already observed many carbonyl groups are ~1700-1730 cm-1 How to distinguish? O R O O R Ketone ~1715 cm-1 for carbonyl R H R OH Aldehyde Acid observe aldehyde C-H stretch observe broad O-H stretch Two peaks between 2700-2900 cm-1 ~3000 cm-1 Small Rings Small rings also have a shift in vibrational frequency to higher energy, Therefore 5,4, or 3-membered rings have the carbonyl stretching frequency shifted O 1715 cm-1 O 1745 cm-1 O 1785 cm-1 Angle strain in these rings causes the carbonyl group to have more electron density, Therefore a “stiffer” bond Infrared Spectroscopy C-N bonds C-N bonds are in similar regions to C-C bonds Wavenumber (cm-1) C N ~1200 cm-1 C N ~1600 cm-1 C N >2200 cm-1 The intensity of absorbance, however is higher due to greater dipole of C-N bond compared to C-C Fingerprint Region The so-called “fingerprint” region is below ~1500 cm-1 Vibrations in this region are often complex and hard to assign to a specific functional group of the molecule -a given molecule, though, has a DISTINCT pattern in this region (reason for this region being called the “fingerprint” region) One common pattern – differentiating substitution isomers Ortho Meta Para CH3 One example: Aromatic isomers one peak 770-735 cm-1 three peaks 900-860, 810-750, 725-680 cm-1 one peak 860-800 cm-1 CH3 CH3 Cl Cl Cl Fingerprint Region CH3 Cl Strong peak 747 cm-1 CH3 Strong peaks 863, 773, 682 cm-1 Cl CH3 Strong peak 806 cm-1 Cl Overtone and Combination Bands Overtone -when assigning IR spectra be careful to note overtone bands (an intense peak will display a smaller peak at a multiple [2x, 3x, etc.] of that peak) Combination Bands Two or more vibrations can couple to cause a vibration at a different position (vibrations must be “coupled” to observe these combination bands) 2nd Overtone ~3430 cm-1 Strong carbonyl stretch ~1715 cm-1 Ultraviolet-Visible Spectroscopy (UV-vis) Another analytical tool to determine organic structures is UV-vis spectroscopy Similar to IR, this is another spectroscopic technique, therefore a sample is irradiated with light of a particular wavelength If the compound absorbs the light, the detector will record the intensity of absorbance In an UV-vis spectrum the light used is between ~200 nm to ~700 nm (UV range is ~200 nm to ~370 nm, while visible light is ~370 nm to 700 nm) Remember that in IR the wavelength of light used was 2.5 – 25 µm (between one to two orders of magnitude larger than UV light) Ultraviolet-Visible Spectroscopy UV light is thus much higher in energy than IR light Instead of causing molecular vibrations, UV-vis light causes electronic excitations An electron is excited from the HOMO to the LUMO Ethylene LUMO E h! Required energy Ethylene HOMO If the correct amount of energy is applied (i.e. the correct wavelength of light), the excitation of one electron from the HOMO to the LUMO will occur Ultraviolet-Visible Spectroscopy The amount of energy required is thus the energy gap between the HOMO and LUMO As the HOMO-LUMO gap changes, the wavelength required for excitation changes (remember that a lower wavelength is higher in energy) The HOMO-LUMO energy gap is affected by the amount of conjugation A conjugated diene system has a lower HOMO-LUMO energy gap than an isolated double bond Therefore a more conjugated system has a higher wavelength of absorbance (higher wavelength is lower in energy, therefore smaller energy gap) Ultraviolet-Visible Spectroscopy Information Obtained from a UV-vis The major piece of information is the point of maximum absorbance (called λmax) In addition an absorbance is characterized by how strongly the molecule absorbs Beer’s law: A = ε•c•l c = concentration of sample l = path length of sample ε = molar absorbtivity (extinction coefficient) *characteristic of sample If the molecule absorbs more strongly, it has a higher ε Ultraviolet-Visible Spectroscopy As seen earlier, the position of the λmax indicates the HOMO-LUMO energy gap A more conjugated system has a lower HOMO-LUMO energy gap, therefore the λmax will be of a longer wavelength λmax 171 nm 180 nm 227 nm Only conjugated alkenes will cause a shift in λmax Alkyl substitution causes a shift of ~5 nm, but conjugation causes shift of ~30 nm As the conjugation increases, the shift will increase Compounds that are colored to our eye therefore must be very conjugated (the lowest wavelength human eyes detect is ~370 nm) Ultraviolet-Visible Spectroscopy Organic colored compounds: β-carotene (λmax = 453 and 483 nm) CO2 Br Br O O Br O Br Eosin Y (λmax = 517 nm)