Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Thermomechanical analysis wikipedia , lookup
Hydrogen-bond catalysis wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Thermodynamics wikipedia , lookup
Electrochemistry wikipedia , lookup
Marcus theory wikipedia , lookup
Click chemistry wikipedia , lookup
Self-assembly of nanoparticles wikipedia , lookup
Chemical reaction wikipedia , lookup
Lewis acid catalysis wikipedia , lookup
Allotropes of carbon wikipedia , lookup
Stoichiometry wikipedia , lookup
Electrolysis of water wikipedia , lookup
George S. Hammond wikipedia , lookup
Transition state theory wikipedia , lookup
Bioorthogonal chemistry wikipedia , lookup
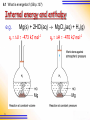
6 1 Energetics 6.1 What is Energetics? 6.2 Enthalpy Changes Related to Breaking and Forming of Bonds 6.3 Standard Enthalpy Changes 6.4 Experimental Determination of Enthalpy Changes by Calorimetry 6.5 Hess’s Law 6.6 Calculations involving Standard Enthalpy Changes of Reactions 6.1 What is energetics? (SB p.136) What is energetics? Energetics is the study of energy changes associated with chemical reactions. Thermochemistry is the study of heat changes associated with chemical reactions. 2 Internal Energy (U) U = kinetic energy + potential energy 3 Kinetic Energy T (K) translational rotational vibrational heat Energy Potential Energy Relative position among particles Bond breaking P.E. Bond forming P.E. 4 Bond breaking : - H – H(g) H(g) + H(g) P.E. Bond forming : - H(g) + H(g) H – H(g) Ionization : Na(g) Na+(g) + e 5 P.E. P.E. Q.1 4H(g) + 2O(g) Internal energy U1 bond breaking bond forming 2H2(g) + O2(g) U = U2 – U1 = -(y-x) kJ U2 2H2O(l) Reaction coordinate 6 6.1 What is energetics? (SB p.137) Internal energy and enthalpy H = U + PV enthalpy 7 Internal energy 6.1 What is energetics? (SB p.137) Internal energy and enthalpy e.g. Mg(s) + 2HCl(aq) MgCl2(aq) + H2(g) qv = U = -473 kJ mol1 Mg 8 qp = H = -470 kJ mol1 Mg qv = U = -473 kJ mol1 qv = qp – 3 = qp – w = qp - PV qp = H = -470 kJ mol1 PV (Nm2)(m3) = Nm Force displacement Work done against the surroundings 9 6.1 What is energetics? (SB p.138) Internal energy and enthalpy H = U + PV = U + PV at constant P qp = qv Heat Heat change at change at fixed P fixed V 10 + PV Work done 6.1 What is energetics? (SB p.138) Internal energy and enthalpy qp = qv + PV On expansion, PV > 0 Work done by the system against the surroundings System gives out less energy to the surroundings qp is less negative than qv (less exothermic) 11 6.1 What is energetics? (SB p.138) Internal energy and enthalpy qp = qv + PV On contraction, PV < 0 Work done by the surroundings against the system System gives out more energy to the surroundings qp is more negative than qv (more exothermic) 12 • H is more easily measured than H as most reactions happen in open vessels. i.e. at constant pressure. • The absolute values of H and H cannot be measured. 13 6.1 What is energetics? (SB p.138) Exothermic and endothermic reactions An exothermic reaction is a reaction that releases heat energy to the surroundings. (H = -ve) 14 6.1 What is energetics? (SB p.139) Check Point 6-1 Exothermic and endothermic reactions An endothermic reaction is a reaction that absorbs heat energy from the surroundings. (H = +ve) 15 6.1 What is energetics? (SB p.136) Law of conservation of energy The law of conservation of energy states that energy can neither be created nor destroyed, but can be exchanged between a system and its surroundings 16 Exothermic : P.E. of the system K.E. of the surroundings Endothermic : K.E. of the surroundings P.E. of the system 17 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.140) Enthalpy changes related to breaking and forming of bonds CH4 + 2O2 CO2 + 2H2O 18 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.140) In an exothermic reaction, E absorbed to break bonds < E released as bonds are formed. 19 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.140) Enthalpy changes related to breaking and forming of bonds N2(g) + 2O2(g) 2NO2(g) 20 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.140) In an endothermic reaction, E absorbed to break bonds > E released as bonds are formed. Check Point 6-2 21 For non-gaseous reactions, PV 0 H = U + PV U For gaseous reactions, H = U + PV = U + (n)RT (PV = nRT) 22 Q.2 CH4(g) + 2O2(g) CO2(g) + 2H2O(l) Given : R = 8.314 J K1 mol1, T = 298 K U = H – (n)RT 8.314 890 (1 3) 298 1000 = 885 kJ mol1 23 C(g) + 4H(g) + 4O(g) Enthalpy H1 bond breaking bond forming CH4(g) + 2O2(g) H = 890 kJ mol1 H2 CO2(g) + 2H2O(l) Reaction coordinate 24 At 298K CH4(g) + 2O2(g) CO2(g) + 2H2O(l) At 373K CH4(g) + 2O2(g) CO2(g) + 2H2O(g) 2H2O(l) 373K 25 2H2O(g) 373K +88 kJ H / kJ mol1 890 802 C(g) + 4H(g) + 4O(g) Enthalpy H1 CH4(g) + 2O2(g) 298 K H = 890 kJ mol1 H2 CO2(g) + 2H2O(l) Reaction coordinate 26 C(g) + 4H(g) + 4O(g) Enthalpy H 1’ CH4(g) + 2O2(g) 373 K H = 890 kJ mol1 H2’ CO2(g) + 2H2O(l) Assume constant H Reaction coordinate 27 C(g) + 4H(g) + 4O(g) Enthalpy CH4(g) + 2O2(g) 373 K H = 890 kJ mol1 CO2(g) + 2H2O(l) In fact, H depends on T Reaction coordinate 28 C(g) + 4H(g) + 4O(g) Enthalpy CH4(g) + 2O2(g) 373 K H = 802 kJ mol1 CO2(g) + 2H2O(g) CO2(g) + 2H2O(l) H = +88 kJ mol1 Reaction coordinate 29 6.3 Standard Enthalpy Changes 30 6.3 Standard enthalpy changes (SB p.141) Standard enthalpy changes CH4(g) + 2O2(g) CO2(g) + 2H2O(g) H = -802 kJ mol-1 at 373 K CH4(g) + 2O2(g) CO2(g) + 2H2O(l) H = -890 kJ mol-1 at 298 K 31 6.3 Standard enthalpy changes (SB p.141) Standard enthalpy changes As enthalpy changes depend on temperature and pressure, it is necessary to define standard conditions: 1. 2. 3. elements or compounds in their normal physical states; a pressure of 1 atm (101325 Nm-2); and a temperature of 25oC (298 K) Enthalpy change under standard conditions denoted by symbol: H 32 Standard enthalpy change of reaction The enthalpy change when the molar quantities of reactants as stated in the equation react under standard conditions. 2H2(g) + O2(g) 2H2O(l) H = 572 kJ mol1 33 per mole of O2 Standard enthalpy change of reaction 2H2(g) + O2(g) 2H2O(l) H = 572 kJ mol1 per mole of O2 1 H2(g) + O2(g) H2O(l) 2 H = 286 kJ mol1 per mole of H2 or H2O 4H2(g) + 2O2(g) 4H2O(l) H = 1144 kJ H depends on the equation 34 Standard enthalpy change of formation Hf The enthalpy change when one mole of the substance is formed from its elements under standard conditions. 1 H2(g) + O2(g) H2O(l) 2 35 Hf [H2O] = 286 kJ mol1 Q.3 Hf [element] = 0 kJ mol1 O2(g) O2(g) Hf [O2] = 0 kJ mol1 C(graphite) C(diamond) Most stable allotrope 36 Hf [diamond] = +1.9 kJ mol1 Q.4 (i) C(graphite) + O2(g) CO2(g) (ii) C(graphite) + 2H2(g) CH4(g) (iii) 1 Mg(s) + 2 O2(g) MgO(s) 1 3 Na(s) + 2 H2(g) + C(graphite) + O2(g) NaHCO3(s) 2 (iv) (v) 37 2C(graphite) + 2H2(g) + O2(g) CH3COOH(l) Q.5 1 H2(g) + O2(g) H2O(l) 2 qv = U = 140.3 kJ per g of H2 no.of moles of H2 (g) used 1.00 0.496 mol 2 1.008 0.496 no.of moles of O2 (g) used mol 0.248 mol 2 n = 0 – 0.496 – 0.248 = -0.744 mol 38 Q.5 H = U + nRT 8.31 kJ mol -1 K -1 (298 K) 140.3 kJ (-0.744 mol) 1000 = -142.1 kJ Heat released for the formation of 0.496 mol of water 142.1 kJ -1 -286.5 kJ mol Molar Hf[H2O] = 0.496 mol 39 Standard enthalpy change of combustion Hc The enthalpy change when one mole of the substance undergoes complete combustion under standard conditions. C2H5OH(l) + 3O2(g) 2CO2(g) + 3H2O(l) Hc [C2H5OH(l)] = -1368 kJ mol1 40 6.3 Standard enthalpy changes (SB p.147) Substance Hc (kJ mol-1) C (diamond) C (graphite) -395.4 -393.5 Enthalpy C(diamond) + O2(g) 1.9 395.4 C(graphite) + O2(g) 393.5 CO2(g) Reaction coordinate 41 Hf [diamond] = +1.9 kJ mol1 Q.6 (a) 1 C(graphite) + O2(g) CO(g) 2 Incomplete combustion H = Hf [CO(g)] Hc [graphite] (b) 2H2(g) + O2(g) 2H2O(l) H = 2 Hc [H2(g)] = 2 Hf [H2O(l)] 42 Q.6 (c) C(graphite) + O2(g) CO2(g) H = Hc [graphite] = Hf [CO2(g)] (d) CH4(g) + 2O2(g) CO2(g) + 2H2O(l) H = Hc [CH4(g)] Hf [CO2(g)] 2 Hf [H2O(l)] Not formed from elements Check Point 6-3 43 6.3 Standard enthalpy changes (SB p.142) Standard enthalpy changes of neutralization Standard enthalpy change of neutralization (Hneut) is the enthalpy change when one mole of water is formed from the neutralization of an acid by an alkali under standard conditions. e.g. H+(aq) + OH-(aq) H2O(l) Hneut = -57.3 kJ mol-1 44 6.3 Standard enthalpy changes (SB p.142) Standard enthalpy changes of neutralization Enthalpy level diagram for the neutralization of a strong acid and a strong alkali 45 6.3 Standard enthalpy changes (SB p.142) Acid HCl HCl HCl HF NH3(aq) + H2O(l) Alkali NaOH KOH NH3 NaOH Hneu -57.1 -57.2 -52.2 -68.6 NH4+(aq) + OH(aq) H1 > 0 H+(aq) + OH-(aq) + Cl(aq) H2O(l) + Cl(aq) H2 = -57.3 NH3(aq) + HCl(aq) NH4Cl (aq) Hneu = H1 + H2 = 52.2 kJ mol1 46 6.3 Standard enthalpy changes (SB p.142) Acid HCl HCl HCl HF HF(aq) Hneu -57.1 -57.2 -52.2 -68.6 Alkali NaOH KOH NH3 NaOH H+(aq) + F(aq) H1 < 0 H+(aq) + OH-(aq) + Na+(aq) H2O(l) + Na+(aq) H2 = -57.3 HF(aq) + NaOH(aq) NaF(aq) + H2O(l) Hneu = H1 + H2 = 68.6 kJ mol1 47 6.3 Standard enthalpy changes (SB p.142) Standard enthalpy change of solution Standard enthalpy change of solution (Hsoln) is the enthalpy change when one mole of a solute is dissolved in a specified number of moles of solvent (e.g. water) under standard conditions. NaCl(s) + 10H2O(l) Na+(aq) + Cl-(aq) Hsoln[NaCl(s)]= +2.008 kJ mol-1 NaCl(aq) 48 dilution NaCl(aq) H > 0 6.3 Standard enthalpy changes (SB p.142) Standard enthalpy change of solution Standard enthalpy change of solution (Hsoln) is the enthalpy change when one mole of a solute is dissolved to form an infinitely dilute solution under standard conditions. concentration 0 NaCl(s) + water Na+(aq) + Cl-(aq) Hsoln= +4.98 kJ mol-1 49 6.3 Standard enthalpy changes (SB p.143) Standard enthalpy change of solution e.g. NaCl(s) + water Na+(aq) + Cl-(aq) Hsoln= +4.98 kJ mol-1 + 4.98 kJ mol1 Enthalpy level diagram for the dissolution of NaCl 50 6.3 Standard enthalpy changes (SB p.143) Standard enthalpy change of solution e.g. LiCl(s) + water Li+(aq) + Cl-(aq) Hsoln= -37.2 kJ mol-1 Enthalpy level diagram for the dissolution of LiCl in water 51 6.3 Standard enthalpy changes (SB p.143) Standard enthalpy change of solution 52 Salt Hsoln(kJ mol-1) NH3 NaOH HCl H2SO4 LiCl NaCl NaNO3 NH4Cl 35.5 43.1 73.0 74.0 37.2 +4.98 +21.0 +22.6 6.4 Experimental Determination of Enthalpy Changes by Calorimetry 53 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.148) Experimental determination of enthalpy changes by calorimetry Calorimeter is any set-up used for the determination of H. By temperature measurement. H = qp = (m1c1 + m2c2)T 54 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.148) H = qp = (m1c1 + m2c2)T where m1 is the mass of the reaction mixture, m2 is the mass of the calorimeter, c1 is the specific heat capacity of the reaction mixture, c2 is the specific heat capacity of the calorimeter, T is the temperature change of the reaction mixture. 55 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.149) Determination of enthalpy change of neutralization 56 If the reaction is fast enough, T1 T2 T1 T2 H (m1c1 + m2c2)(T2 – T0) H = (m1c1 + m2c2)(T1 – T0) T0 t1 57 t2 Check Point 6-4(a) 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.150) Determination of enthalpy change of combustion The Philip Harris calorimeter used for determining the enthalpy change of combustion of a liquid fuel 58 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.151) Determination of enthalpy change of combustion A simple apparatus used to determine the enthalpy change of combustion of ethanol 59 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.151) Heat evolved = (m1c1 + m2c2) ΔT Where m1 is the mass of water in the calorimeter, m2 is the mass of the calorimeter, c1 is the specific heat capacity of the water, c2 is the specific heat capacity of the calorimeter, ΔT is the temperature change of the reaction 60 Q.7(Example) qp (800 g)(4.18 J g-1K1 )(13 K) 43.5 kJ 1.5 g no. of moles of ethanol 0.326 mol 46.0 g ΔHc [ethanol] - 43.5 kJ - 1330 kJ mol -1 0.0326 mol heat given out Check Point 6-4(c) 61 6.5 Hess’s Law 62 6.5 Hess’s law (SB p.153) Hess’s Law Hess’s law of constant heat summation states that the total enthalpy change accompanying a chemical reaction is independent of the route by which the chemical reaction takes place and depends only on the difference between the total enthalpy of the reactants and that of the products. 63 6.5 Hess’s law (SB p.153) Hess’s Law H2 A(HA) Route 2 C Route 1 H1 H4 H3 B(HB) H5 D Route 3 H1 = HB – HA = H2 + H3 = H4 + H5 64 6.5 Hess’s law (SB p.155) Importance of Hess’s law The enthalpy change of some chemical reactions cannot be determined directly because: • • • the reactions cannot be performed/controlled in the laboratory the reaction rates are too slow the reactions may involve the formation of side products But the enthalpy change of such reactions can be determined indirectly by applying Hess’s Law. 65 6.5 Hess’s law (SB p.153) Enthalpy change of formation of CO(g) C(graphite) + ½O2(g) Hf [CO(g)] CO(g) Hf [CO(g)] cannot be determined directly due to further oxidation of CO to CO2 The reaction cannot be controlled. 66 6.5 Hess’s law (SB p.153) Enthalpy change of formation of CO(g) Hc [graphite] = -393 kJ mol-1 Hc [CO(g)] = -283.0 kJ mol-1 C(graphite) + ½O2(g) + ½O2(g) Hf [CO(g)] + ½O2(g) H1 CO2(g) H2 Hf [CO(g)] + H2 = H1 Hf [CO(g)] = H1 - H2 = -393 - (-283 ) = -110 kJ mol-1 67 CO(g) 6.5 Hess’s law (SB p.155) Enthalpy cycle (Born-Haber cycle) • Relate the various equations involved in a reaction C(graphite) + ½O2(g) + ½O2(g) 68 Hf [CO(g)] CO(g) + ½O2(g) H1 CO2(g) H2 6.5 Hess’s law (SB p.153) Steps for drawing Born-Haber cycle 1. Give the equation for the change being considered. C(graphite) + ½O2(g) 69 Hf [CO(g)] CO(g) 6.5 Hess’s law (SB p.153) Steps for drawing Born-Haber cycle 2. Complete the cycle by giving the equations for the combustion reactions of reactants and products. C(graphite) + ½O2(g) + ½O2(g) H1 Hf [CO(g)] CO(g) + ½O2(g) CO2(g) H2 Hf [CO(g)]= Hc[graphite] - Hc[CO(g)] 70 6.5 Hess’s law (SB p.153) Steps for drawing Born-Haber cycle 2. Complete the cycle by giving the equations for the combustion reactions of reactants and products. C(graphite) + ½O2(g) + ½O2(g) H1 Hf [CO(g)] CO(g) + ½O2(g) CO2(g) H2 Hf [CO(g)]= Hc[reactant] - Hc[product] 71 Calculation of standard enthalpy change of formation from standard enthalpy changes of combustion 6B Hf = Hc [reactants] - Hc [product] 72 Q.8 4C(graphite) + 5H2(g) Hf [C4H10(g)] + 4O2(g) 5Hc [H2(g)] + 2.5 O2(g) 4Hc [graphite] C4H10(g) Hc [C4H10(g)] + 6.5 O2(g) 4CO2(g) + 5H2O(l) Hf [C4H10(g)] = 4Hc[C(graphite)] + 5Hc[H2(g)] - Hc[C4H10(g)] = [4(-393)+ 5(-286) – (2877)] kJ mol1 = 125 kJ mol1 73 Q.8 Method B : By addition and/or subtraction of equations with known Hc Hc /kJ mol1 (1) C(graphite) + O2(g) CO2(g) 1 (2) H2(g) + O2(g) H2O(l) 2 (3) C4H10(g) + 6 393 286 1 O2(g) 4CO2(g) + 5H2O(l) 2877 2 Overall reaction : 4(1) + 5(2) – (3) Hf [C4H10(g)] 4C(graphite) + 5H2(g) C4H10(g) Hf [C4H10(g)] = [4(-393) + 5(-286) (2877)] kJ mol1 = 125 kJ mol1 74 6.5 Hess’s law (SB p.154) Enthalpy level diagram • Relate substances together in terms of enthalpy changes of reactions Enthalpy level diagram for the oxidation of C(graphite) to CO2(g) 75 Steps for drawing enthalpy level diagram Enthalpy / kJ mol1 1. Draw the enthalpy level of elements. 76 C(graphite) + O2(g) Steps for drawing enthalpy level diagram Enthalpy / kJ mol1 2. Enthalpies of elements are arbitrarily taken as zero. 77 C(graphite) + O2(g) Steps for drawing enthalpy level diagram Enthalpy / kJ mol1 3. Higher enthalpy levels are drawn above that of elements 78 C(graphite) + O2(g) Steps for drawing enthalpy level diagram Enthalpy / kJ mol1 4. Lower enthalpy levels are drawn below that of elements C(graphite) + O2(g) Hc[graphite] = 393 kJ CO2(g) Route 1 79 Steps for drawing enthalpy level diagram Enthalpy / kJ mol1 4. Lower enthalpy levels are drawn below that of elements C(graphite) + O2(g) Hf[CO(g)] = 110 kJ Hc[graphite] = 393 kJ 1 O (g) 2 2 Hc[CO(g)] = 283 kJ CO2(g) Route 1 80 CO(g) + Route 2 6.5 Hess’s law (SB p.158) (c) The formation of ethyne (C2H2(g) can be represented by the following equation: 2C(graphite) + H2(g) C2H2(g) (i) Draw an enthalpy cycle relating the above equation to carbon dioxide and water. (ii) Calculate the standard enthalpy change of formation of ethyne. (Given: Hc [C(graphite)] = -393.5 kJ mol-1; Hc [H2(g)] = -285.8 kJ mol-1; Hc [C2H2(g)] = -1299 kJ mol-1) 81 6.5 Hess’s law (SB p.158) Hf 2C(graphite) + H2(g) C2H2(g) 2Hc[graphite] + 2O2(g) Hc[H2(g)] + 0.5O2(g) Hc[C2H2(g)] + 2.5O2(g) 2CO2(g) + H2O(l) By Hess’s law, Hf + Hc[C2H2(g)] = 2Hc[graphite] + Hc[H2(g)] Hf = 2Hc[graphite] + Hc[H2(g)] - Hc[C2H2(g)] = [2(393.5) + (285.8) –(1299)] kJ mol1 = +226.2 kJ mol1 82 Enthalpy / kJ mol1 (iii). Draw an enthalpy level diagram for the reaction using the enthalpy changes in (ii) C2H2(g) + 2.5O2(g) Hf[C2H2(g)] 2C(graphite) + H2(g) + 2.5O2(g) 2Hc[graphite] Hc[C2H2(g)] 2CO2(g) + H2(g) + 0.5O2(g) Hc[H2(g)] 2CO2(g) + H2O(l) Route 1 83 Route 2 6.6 Calculations involving Standard Enthalpy Changes of Reactions 84 6.6 Calculations involving standard enthalpy changes of reactions (SB p.159) Calculation of standard enthalpy change of reaction from standard enthalpy changes of formation H from Hf 85 Q.9 NH3(g) + HCl(g) H Hf[HCl(g)] Hf[NH3(g)] NH4Cl(s) Hf[NH4Cl(s)] 0.5N2(g) + 1.5H2(g) + 0.5H2(g) + 0.5Cl2(g) By Hess’s law, Hf[NH3(g)] + Hf[HCl(g)] + H = Hf[NH4Cl(s)] H = Hf[NH4Cl(s)] - Hf[NH3(g)] - Hf[HCl(g)] = [314 –(46) –(92)] kJ mol1 = 176 kJ mol1 86 Q.9 NH3(g) + HCl(g) H Hf[HCl(g)] Hf[NH3(g)] NH4Cl(s) Hf[NH4Cl(s)] 0.5N2(g) + 1.5H2(g) + 0.5H2(g) + 0.5Cl2(g) By Hess’s law, Hf[NH3(g)] + Hf[HCl(g)] + H = Hf[NH4Cl(s)] H = Hf[NH4Cl(s)] - Hf[NH3(g)] - Hf[HCl(g)] Hreaction = Hf [products] - Hf [reactants] 87 Q.10 4CH3NHNH2(l) + 5N2O4(l) H 5Hf[N2O4(l)] 4Hf[CH3NHNH2(l)] 4CO2(g) + 9N2(g) + 12H2O(l) 4Hf[CO2(g)] 12Hf[H2O(l)] 4C(graphite) + 12H2(g) + 4N2(g) + 5N2(g) + 10O2(g) By Hess’s law, H = [4(393) + 12(286) - 4(+53) – 5(20)] kJ = 5116 kJ Highly exothermic/ignites spontaneously Used as rockel fuel in Apollo 11 88 Q.11 C3H6(g) + H2(g) H C3H8(g) H /kJ mol1 (1) C3H6(g) + 4.5O2(g) 3CO2(g) + 3H2O(l) (2) H2(g) + 0.5O2(g) H2O(l) (3) C3H8(g) + 5O2(g) 3CO2(g) + 4H2O(l) Overall reaction : (1) + (2) – (3) H = [(2058) + (286) – (2220)] kJ mol1 = 124 kJ mol1 89 2058 286 2220 Q.11 C3H6(g) + H2(g) H Hf[H2O(l)] Hc[C3H6(g)] C3H8(g) Hc[C3H8(g)] 3H2O(l) + 3CO2 (g) + H2O(l) By Hess’s law, H + (2220) = (2058) + (286) H = [(2058) + (286) – (2220)] kJ mol1 = 124 kJ mol1 90 Q.11 C3H6(g) + H2(g) H Hc[H2(g)] Hc[C3H6(g)] C3H8(g) Hc[C3H8(g)] 3H2O(l) + 3CO2 (g) + H2O(l) Hf = Hc [reactants] - Hc [product] Hrx = Hc [reactants] - Hc [products] 91 Relative stability of compounds and Hf Hf indicates the energetic stability of the compound with respect to its elements Elements Compound Hf Hf < 0 energetically more stable than its elements Hf > 0 energetically less stable than its elements 92 H2(g) + O2(g) H2O2(l) Hf [H2O2(l)] = 188 kJ mol1 Enthalpy / kJ mol1 H2O2(l) H2O(l) + 0.5O2(g) 93 Hrx = 98 kJ mol1 H2(g) + O2(g) Hf[H2O2(l)] = 188 kJ H2O2(l) Hrx= 98 kJ H2O(l) + 0.5O2(g) H2O2 is energetically stable w.r.t. H2 and O2 Enthalpy / kJ mol1 H2O2 is energetically unstable w.r.t. H2O and 0.5O2 94 H2(g) + O2(g) Hf[H2O2(l)] = 188 kJ H2O2(l) Hrx= 98 kJ H2O(l) + 0.5O2(g) Enthalpy / kJ mol1 H2 and O2 are energetically unstable w.r.t. H2O2 and H2O 95 H2(g) + O2(g) Hf[H2O2(l)] = 188 kJ Hc[H2(g) ] = 286 kJ H2O2(l) Hrx= 98 kJ H2O(l) + 0.5O2(g) C(diamond) C(graphite) H = 2 kJ mol1 Enthalpy / kJ mol1 Energetically unstable 96 C(diamond) + O2(g) H = 2 kJ C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ C(diamond) C(graphite) H = 2 kJ mol1 Enthalpy / kJ mol1 Kinetically The rate of conversion is extremely low stable 97 C(diamond) + O2(g) H = 2 kJ C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ C(g) + O(g) + O(g) Enthalpy / kJ mol1 bond breaking 98 bond forming C(diamond) + O2(g) C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ Rate of reaction depends on the ease of bond breaking in reactants (e.g. diamond) Enthalpy / kJ mol1 The minimum energy required for a reaction to start is known as the activation energy, Ea 99 C(diamond) + O2(g) H = 2 kJ C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ C(diamond) C(graphite) H = 2 kJ mol1 Enthalpy / kJ mol1 The extremely low rate is due to high Ea 100 C(diamond) + O2(g) H = 2 kJ C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ C(diamond) C(graphite) H = 2 kJ mol1 Enthalpy / kJ mol1 Ea is always > 0 as bond breaking is endothermic 101 C(diamond) + O2(g) H = 2 kJ C(graphite) + O2(g) Hc[diamond] = 395 kJ CO2(g) Hc[graphite] = 393 kJ Ea tells how fast a reaction can proceed. Rate of reaction / kinetics / kinetic stability Hf tells how far a reaction can proceed Equilibrium / energetics / energetic stability 102 Hf Energetic stability Hf < 0 higher energetic stability w.r.t. its elements Hf > 0 lower energetic stability w.r.t. its elements Ea kinetic stability (rate of reaction) Higher Ea Lower Ea 103 higher kinetic stability of reactants w.r.t. products lower rate to give products lower kinetic stability of reactants w.r.t. products higher rate to give products diamond graphite Diamond energetically unstable w.r.t. graphite kinetically stable w.r.t. graphite Graphite stable w.r.t. diamond energetically and kinetically 104 diamond graphite diamond 105 extremely slow extremely slow graphite 6.7 Entropy change (SB p.164) 6.7 Spontaneity of Changes 106 Spontaneity : The state of being spontaneous Spontaneous : - self-generated - natural - happening without external influence external external boundary internal external external 107 6.7 Entropy change (SB p.164) A process is said to be spontaneous • If no external “forces” are required to keep the process going, although external “forces” may be required to get the process started (Ea). • The process may be a physical change or a chemical change 108 6.7 Entropy change (SB p.164) • Spontaneous physical change: E.g. condensation of steam at 25°C • Spontaneous chemcial change: E.g. burning of wood once the fire has started Exothermic spontaneous ? Endothermic not spontaneous ? Q.12 109 110 Change Exothermic ? (Yes / No) Spontaneous ? (Yes / No) Burning of CO at 25°C Yes Yes Condensation of steam at 25°C Yes Yes Melting of ice at 25°C No Yes Dissolution of NH4Cl in water at 25°C No Yes 6.7 Entropy change (SB p.164) • Exothermicity is NOT the only reason for the spontaneity of a process • Some spontaneous changes are endothermic • E.g. Melting of ice Dissolution of NH4Cl in water 111 6.7 Entropy change (SB p.164) Melting of ice Dissolution of ammonium nitrate in water 112 6.7 Entropy change (SB p.165) Entropy • Entropy is a measure of the randomness or the degree of disorder (freedom) of a system Solid Liquid Entropy increases 113 Gas 6.7 Entropy change (SB p.166) Entropy change (S) • Entropy change means the change in the degree of disorder of a system S = Sfinal - Sinitial 114 6.7 Entropy change (SB p.166) Positive entropy change (S > 0) • Increase in entropy • Sfinal > Sinitial • Example: Ice (low entropy) Water (high entropy) S = Swater – Sice = +ve 115 6.7 Entropy change (SB p.166) Negative entropy change (S < 0) • Decrease in entropy • Sinitial > Sfianl • Example: Water (high entropy) Ice (low entropy) S = Sice – Swater = -ve Q.13 116 Changes CO(g) + O2(g) CO2(g) H2O(g) H2O(l) H2O(s) H2O(l) NH4Cl(s) NH4Cl(aq) 117 S 118 Changes S 2CO(g) + O2(g) 2CO2(g) -ve H2O(g) H2O(l) -ve H2O(s) H2O(l) +ve NH4Cl(s) NH4Cl(aq) +ve Changes C(s) + O2(g) CO2(g) SO2(g) + O2(g) SO3(g) Diffusion of a drop of ink in water diamond graphite 119 S Changes 2 C(s) + O2(g) 2 CO2(g) 120 S +ve 2SO2(g) + O2(g) 2SO3(g) -ve Diffusion of a drop of ink in water +ve diamond graphite +ve Consider an isolated system which has no exchange of energy and matter with its surroundings 121 S1 S2 S = S2 – S1 > 0 Which one is spontaneous ? S2 S1 S = S1 – S2 < 0 122 S1 S2 Spontaneous, S = S2 – S1 > 0 S2 S1 Not spontaneous, S = S1 – S2 < 0 123 A molecular statistical interpretation Free adiabatic expansion of an ideal gas into a vacuum. vacuum Slit is open Probability 1 1 1 1 1 2 2 2 2 16 124 A molecular statistical interpretation Free adiabatic expansion of an ideal gas into a vacuum. 1 mole vacuum Slit is open Probability 1 2 125 61023 1 1.81023 10 A spontaneous process taking place in an isolated system is always associated with an increase in entropy (I.e. S > 0) In closed system, 6B The spontaneity of a process depends on both H and S. The driving force of a process is a balance of H and S. 126 6.7 Entropy change (SB p.166) Ice (less entropy) Water (more entropy) H is +ve not favourable S is +ve favourable Considering both S & H , the process is spontaneous Q.14 127 Changes H2O(g) H2O(l) at 25°C H2O(g) H2O(l) at 110°C H2O(s) H2O(l) at 25°C H2O(s) H2O(l) at -10°C 128 H S Spontaneous (Yes / No) 129 Changes H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Favourable Changes 130 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Not favourable Changes 131 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Spontaneity depends on temperature Changes 132 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Favourable Changes 133 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Not favourable Changes 134 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Spontaneity depends on temperature Changes 135 H S Spontaneous (Yes / No) H2O(g) H2O(l) at 25°C -ve -ve Yes H2O(g) H2O(l) at 110°C -ve -ve No H2O(s) H2O(l) at 25°C +ve +ve Yes H2O(s) H2O(l) at -10°C +ve +ve No Spontaneity of a process depends on H, S & T G = H –TS G is the (Gibbs’) free energy J. Willard Gibbs (1839 – 1903) 136 G = H –TS Spontaneity depends on G ‘Free’ means the energy free for work 137 A spontaneous process is always associated with a decrease in the free energy of the system. G < 0 spontaneous process G > 0 not spontaneous process Q.15 138 G = H –TS 139 H S T +ve +ve high +ve +ve low -ve -ve high -ve -ve low G Results G = H –TS 140 H S T G Results +ve +ve high -ve Spontaneous +ve +ve low +ve Not spontaneous -ve -ve high +ve Not spontaneus -ve -ve low -ve Spontaneous G = H –TS 141 H S T -ve +ve high -ve +ve low +ve -ve high +ve -ve low G Results G = H –TS H S T -ve +ve high -ve +ve low +ve -ve high +ve 142 -ve low G Results -ve Spontaneous +ve Not spontaneous G = H –TS Q.16 143 Diamond Graphite H < 0 S > 0 G = H –TS < 0 The process is spontaneous, although activation energy is required to start the conversion. 144 S1 S2 Spontaneous S = S2 – S1 > 0 The entropy of a system can be considered as a measure of the availability of the system to do work. Before expansion, the system is available to do work. After expansion, the system is not available to do work. 145 S1 S2 Spontaneous S = S2 – S1 > 0 The lower the entropy of a system(before expansion), the more available is the system to do work. Thus, entropy is considered as a measure of the useless energy of a system. 146 G = H –TS G = H – TS H = G + TS Total energy Useless energy Useful energy 147 If the universe is an isolated system H is a constant and H is always zero H = G + TS Total energy Useless energy Useful energy 148 cosmic background radiation = 4K Useful Useless energy energy H = G + TS H = G + TS = 0 = G + TS = 0 S always > 0, S always , useless energy always G always < 0, G always , useful energy always 149 In an isolated system, entropy will only increase with time, it will not decrease with time. The second law of thermodynamics 150 If the universe is an isolated system, Suniverse = Ssystem + Ssurroundings > 0 the total entropy (randomness) of the universe will tend to increase to a maximum; the total free energy of the universe will tend to decrease to a minimum. 151 As time increases, the universe will always become more disordered. Entropy is considered as a measure of time. Entropy can be used to distinguish between future and past. 152 Time can only proceed in one direction that results in an increase in the total entropy of the universe. This is known as the arrow of time. 153 The history of the universe minimum entropy, maximum free energy (singularity) H = G +TS T 1.41032 K expanding maximum entropy, minimum free energy 154 Big bang Big chill T0K Planck’s units Planck’s temperature Planck’s length Planck’s time 155 h = Planck’s constant G = gravitational constant c = speed of light in vacuum k = Boltzmann constant hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s units Planck’s temperature Planck’s length Planck’s time 156 Absolute hot beyond which all physical laws break down hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s units Planck’s temperature hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s length hG lP 3 2c 1.616252(81) × 10−35 m Planck’s time 157 Physical significance not yet known Planck’s units Planck’s temperature hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s length hG lP 3 2c 1.616252(81) × 10−35 m Planck’s time 158 1 20 the diameter of proton 10 Planck’s units Planck’s temperature hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s length hG lP 3 2c 1.616252(81) × 10−35 m Planck’s time 159 = 7.310-37 m 22 times of Mr Chio’s wavelength Planck’s units Planck’s temperature hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s length hG lP 2c 3 1.616252(81) × 10−35 m hG tp 2c 5 5.39124(27) × 10−44 s Planck’s time 160 It is the time required for light to travel, in a vacuum, a distance of 1 Planck length. Planck’s units 10-15 s femtosecond(飛秒) 10-18 s attosecond(阿托秒) Time taken for light to travel the length of 3 H atoms Planck’s temperature hc 5 TP 2 2Gk 1.416785(71) × 1032 K Planck’s length hG lP 2c 3 1.616252(81) × 10−35 m hG tp 2c 5 5.39124(27) × 10−44 s Planck’s time 161 Q.17(a) The drop in temperature of the system is accompanied by the rise in temperature of its surroundings. Ssystem < 0 Ssurroundings > 0 Suniverse = Ssystem + Ssurroundings > 0 162 Q.17(b) The drop in entropy of the system is at the cost of the rise in entropy of its surroundings. Ssystem < 0 Ssurroundings > 0 Suniverse = Ssystem + Ssurroundings > 0 163 6.8 Free energy change (SB p.170) Check Point 6-8 164 The END 165 6.1 What is energetics? (SB p.140) Back State whether the following processes are exothermic or endothermic. (a) Melting of ice. (a) Endothermic (b) Dissolution of table salt. (c) Condensation of steam. 166 (b) Endothermic (c) Exothermic Answer 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.141) (a) State the difference between exothermic and endothermic reactions with respect to (i) the sign of H; (ii) the heat change with the surroundings; Answer (iii) the total enthalpy of reactants and products. (a) (i) Exothermic reactions: H = -ve; endothermic reactions: H = +ve (ii) Heat is given out to the surroundings in exothermic reactions whereas heat is taken in from the surroundings in endothermic reactions. 167 (iii) In exothermic reactions, the total enthalpy of products is less than that of the reactants. In endothermic reactions, the total enthalpy is greater than that of the reactants. 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.141) (b) Draw an enthalpy level diagram for a reaction which is (i) endothermic, having a large activation energy. (ii) exothermic, having a small activation energy. Answer 168 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.141) (b) (i) 169 6.2 Enthalpy changes related to breaking and forming of bonds (SB p.141) Back (ii) 170 6.3 Standard enthalpy changes (SB p.147) (a) Why must the condition “burnt completely in oxygen” be emphasized in the definition of standard enthalpy change of combustion? (a) If the substance is not completely burnt in excess oxygen, other products such as C(s) and CO(g) may be formed. The enthalpy change of combustion measured will not be accurate. 171 Answer 6.3 Standard enthalpy changes (SB p.147) (b) The enthalpy change of the following reaction under standard conditions is –566.0 kJ. 2CO(g) + O2(g) 2CO2(g) What is the standard enthalpy change of combustion of carbon monoxide? Answer (b) Standard enthalpy change of combustion of CO 1 = (-566.0) kJ 2 = -283.0 kJ 172 6.3 Standard enthalpy changes (SB p.147) (c) What terms may be given for the enthalpy change of the following reaction? 1 N2(g) + O2(g) NO2(g) 2 (c) ½ Enthalpy change of combustion of nitrogen or enthalpy change of formation of nitrogen dioxide. Back 173 Answer 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.149) Determine the enthalpy change of neutralization of 25 cm3 of 1.25 M hydrochloric acid and 25 cm3 of 1.25 M sodium hydroxide solution using the following data: Mass of calorimeter = 100 g Initial temperature of acid = 15.5 oC (288.5 K) Initial temperature of alkali = 15.5 oC (288.5 K) Final temperature of the reaction mixture = 21.6 oC (294.6 K) The specific heat capacities of water and calorimeter are 4200 J kg-1 K-1 and 800 J kg-1 K-1 respectively. Answer 174 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.149) Assume that the density of the reaction mixture is the same as that of water, i.e. 1 g cm-3. Mass of the reaction mixture = (25 + 25) cm3 1 g cm-3 = 50 g = 0.05 kg Heat given out = (m1c1 + m2c2) T = (0.05 kg 4200 J kg-1 K-1 + 0.1 kg 800 J kg-1 K-1) (294.6 – 288.5) K = 1769 J H+(aq) + OH-(aq) H2O(l) Number of moles of HCl = 1.25 mol dm-3 25 10-3 dm3 = 0.03125 mol Number of moles of NaOH = 1.25 mol dm-3 25 10-3 dm3 = 0.03125 mol Number of moles of H2O formed = 0.03125 mol 175 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.149) Heat given out per mole of H2O formed 1769 J = 0.03125 mol = 56608 J mol-1 The enthalpy change of neutralization is –56.6 kJ mol-1. Back 176 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.151) Determine the enthalpy change of combustion of ethanol using the following data: Mass of spirit lamp before experiment = 45.24 g Mass of spirit lamp after experiment = 44.46 g Mass of water in copper calorimeter = 50 g Mass of copper calorimeter without water = 380 g Initial temperature of water = 18.5 oC (291.5 K) Final temperature of water = 39.4 oC (312.4 K) The specific heat capacities of water and copper calorimeter are 4200 J kg-1 K-1 and 2100 J kg-1 K-1 respectively. 177 Answer 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.151) Heat evolved by the combustion of ethanol = Heat absorbed by the copper calorimeter = (m1c1 + m2c2) T = (0.05 kg 4200 J kg-1 K-1 + 0.38 kg 2100 J kg-1 K-1) (312.4 – 291.5)K = 21067 J C2H5OH(l) + 3O2(g) 2CO2(g) + 3H2O(l) Mass of ethanol burnt = (45.24 – 44.46) g = 0.78 g 0.78 g Number of moles of ethanol burnt = = 0.017 mol 1 46.0 g mol 178 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.151) Heat given out per mole of ethanol 21067 J = 0.017 mol = 1239235 J mol-1 Back = 1239 kJ mol-1 The enthalpy change of combustion of ethanol is –1239 kJ mol-1. There was heat loss by the system to the surroundings, and incomplete combustion of ethanol might occur. Also, the experiment was not carried out under standard conditions. Therefore, the experimentally determined value (-1239 kJ mol-1) is less than the theoretical value of the standard enthalpy change of combustion of ethanol (-1371 kJ mol-1). 179 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.152) 0.02 mol of anhydrous ammonium chloride was added to 45 g of water in a polystyrene cup to determine the enthalpy change of solution of anhydrous ammonium chloride. It is found that there was a temperature drop from 24.5 oC to 23.0 oC in the solution. Given that the specific heat capacity of water is 4200 J kg-1 K-1 and NH4Cl(s) + aq NH4Cl(aq) Calculate the enthalpy change of solution of anhydrous ammonium chloride. (Neglect the specific heat capacity of the polystyrene cup.) Answer 180 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.152) Heat absorbed = m1c1T ( c2 0) = 0.045 kg 4200 J kg-1 K-1 (297.5 – 296) K = 283.5 J (0.284 kJ) Heat absorbed per mole of ammonium chloride = 0.284 kJ 0.02 mol = 14.2 kJ mol-1 The enthalpy change of solution of anhydrous ammonium chloride is +14.2 kJ mol-1. Back 181 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) (a) A student tried to determine the enthalpy change of neutralization by putting 25.0 cm3 of 1.0 M HNO3 in a polystyrene cup and adding 25.0 cm3 of 1.0 M NH3 into it. The temperature rise recorded was 3.11 oC. Given that the mass of the polystyrene cup is 250 g, the specific heat capacities of water and the polystyrene cup are 4200 J kg-1 K-1 and 800 J kg-1 K-1 respectively. Determine the enthalpy change of neutralization of nitric acid and aqueous ammonia. (Density of water = 1 g cm-3) Answer 182 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) (a) Heat evolved = m1c1T + m2c2 T = 0.050 kg 4200 J kg-1 K-1 3.11 K + 0.25 kg 800 J kg-1 K-1 3.11 K = (653.1 + 622) J = 1275.1 J No. of moles of HNO3 used = 1.0 M 25 10-3 dm3 = 0.025 mol 183 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) Back (a) No. of moles of NH3 used = 1.0 M 25 10-3 dm3 = 0.025 mol No. of moles of H2O formed = 0.025 mol Heat evolved per mole of H2O formed 1275.1 J = 0.025 mol = 51.004 kJ mol-1 The enthalpy change of neutralization of nitric acid and aqueous ammonia is –51.004 kJ mol-1. 184 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) (b) When 0.05 mol of silver nitrate was added to 50 g of water in a polystyrene cup, a temperature drop of 5.2 oC was recorded. Assuming that there was no heat absorption by the polystyrene cup, calculate the enthalpy change of solution of silver nitrate. (Specific heat capacity of water = 4200 J kg-1 K-1) Answer 185 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) Back (b) Energy absorbed = mcT = 0.05 kg 4200 J kg-1 K-1 5.2 K = 1092 J No. of moles of AgNO3 used = 0.05 mol 1092 J 0.05 mol = 21.84 kJ mol-1 Energy absorbed per mole of AgNO3 used = The enthalpy change of solution of silver nitrate is +21.84 kJ mol-1. 186 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) (c) A student used a calorimeter as shown in Fig. 6-15 to determine the enthalpy change of combustion of methanol. In the experiment, 1.60 g of methanol was used and 50 g of water was heated up, raising the temperature by 33.2 oC. Given that the specific heat capacities of water and copper calorimeter are 4200 J kg-1 K-1 and 2100 J kg-1 K-1 respectively and the mass of the calorimeter is 400 g, calculate the enthalpy change of combustion of methanol. Answer 187 6.4 Experimental determination of enthalpy changes by calorimetry (SB p.153) (c) Heat evolved = m1c1T + m2c2 T = 50 g 4.18 J g-1 K-1 33.2 K + 400g 2.10 J g-1 K-1 33.2 K = (6939 + 27888) J = 34827 J 1.60 g No. of moles of methanol used = 32.0 g mol -1 = 0.05 mol 34827 J Heat evolved per mole of methanol used = 0.05 mol = 696.5 kJ mol-1 The enthalpy change of combustion of methanol is –696.5 kJ mol-1. 188 6.5 Hess’s law (SB p.158) (a) Given the following thermochemical equation: 2H2(g) + O2(g) 2H2O(l) (i) Is the reaction endothermic or exothermic? (ii) What is the enthalpy change for the following reactions? (1) 2H2O(l) 2H2(g) + O2(g) 1 (2) H2(g) + O2(g) H2O(l) 2 (iii) If the enthalpy change for the reaction H2O(l) H2O(g) is +41.1 kJ mol-1, calculate the H for 2H2(g) + O2(g) 2H2O(g). Answer 189 6.5 Hess’s law (SB p.158) (a) (i) Exothermic (ii) (1) +571.6 kJ mol-1 (2) –285.8 kJ mol-1 (iii) H = [-571.6 + 2 (+41.1)] kJ mol-1 = -489.4 kJ mol-1 190 6.5 Hess’s law (SB p.158) (b) Given the following information about the enthalpy change of combustion of allotropes of carbon: Hc [C(graphite)] = -393.5 kJ mol-1 Hc [C(diamond)] = -395.4 kJ mol-1 (i) Which allotrope of carbon is more stable? (ii) What is the enthalpy change for the following process? C(graphite) C(diamond) 191 Answer 6.5 Hess’s law (SB p.158) (b) (i) Graphite (ii) H = [-393.5 – (-395.4)] kJ mol-1 = +1.9 kJ mol-1 192 6.5 Hess’s law (SB p.158) (c) The formation of ethyne (C2H2(g) can be represented by the following equation: 2C(graphite) + H2(g) C2H2(g) (i) Draw an enthalpy level diagram relating the above equation to carbon dioxide and water. (ii) Calculate the standard enthalpy change of formation of ethyne. (Given: Hc [C(graphite)] = -393.5 kJ mol-1; Hc [H2(g)] = -285.8 kJ mol-1; Hc [C2H2(g)] = -1299 kJ mol-1) 193 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.159) Given the following information, find the standard enthalpy change of the reaction: C2H4(g) + H2(g) C2H6(g) Hf [C2H4(g)] = +52.3 kJ mol-1 Hf [C2H6(g)] = -84.6 kJ mol-1 194 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.159) Back Note: H1 = [Hf (reactants)] = Hf [C2H4(g)] + Hf [H2(g)] H2 = [Hf (products)] = Hf [C2H6(g)] Applying Hess’s law, H1 + H = H2 H = H2 - H1 = Hf [C2H6(g)] – (Hf [C2H4(g)] + Hf [H2(g)]) = [-84.6 – (+52.3 + 0)] kJ mol-1 =-136.9 kJ mol-1 The 195 standard enthalpy change of the reaction is –136.9 kJ mol-1. 6.6 Calculations involving standard enthalpy changes of reactions (SB p.160) Given the following information, find the standard enthalpy change of the reaction: 6PbO(s) + O2(g) 2Pb3O4(s) Hf [PbO(g)] = -220.0 kJ mol-1 Hf [Pb3O4(g)] = -737.5 kJ mol-1 196 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.160) Back Note: H1 = [Hf (reactants)] = 6 Hf [PbO(s)] + Hf [O2(g)] H2 = [Hf (products)] = 2 Hf [Pb3O4(s)] Applying Hess’s law, H1 + H = H2 H = H2 - H1 = 2 Hf [Pb3O4(s)] – (6 Hf [PbO(s)] + Hf [O2(g)]) = [2 (-737.5) – 6 (-222.0) – 0] kJ mol-1 =-155.0 kJ mol-1 The 197 standard enthalpy change of the reaction is –155.0 kJ mol-1. 6.6 Calculations involving standard enthalpy changes of reactions (SB p.160) Given the following information, find the standard enthalpy change of the reaction: Fe2O3(s) + 3CO(g) 2Fe(s) + 3CO2(g) Hf [Fe2O3(s)] = -822.0 kJ mol-1 Hf [CO(g)] = -110.5 kJ mol-1 Hf [CO2(g)] = -393.5 kJ mol-1 198 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.160) Back H1 = [Hf (reactants)] = Hf [Fe2O3(s)] + 2 Hf [CO(g)] H2 = [Hf (products)] = 2 Hf [Fe(s)] + 3 Hf [CO2(g)] Applying Hess’s law, H1 + H = H2 H = H2 - H1 = 2 Hf [Fe(s)] + 3 Hf [CO2(g)] - Hf [Fe2O3(s)] - 3 Hf [CO(g)] = [2 (0) + 3 (-393.5) –(-822.0) – 3 (-110.5)] kJ mol-1 =-27.0 kJ mol-1 199 The standard enthalpy change of the reaction is –27.0 kJ mol-1. Note: 6.6 Calculations involving standard enthalpy changes of reactions (SB p.161) Given the following information, find the standard enthalpy change of the reaction: 4CH3 · NH · NH2(l) + 5N2O4(l) 4CO2(g) + 12H2O(l) + 9N2(g) Hf [CH3 · NH · NH2(l)] = +53 kJ mol-1 Hf [N2O4(l)] = -20 kJ mol-1 Hf [CO2(g)] = -393.5 kJ mol-1 Hf [H2O(l)] = -285.8 kJ mol-1 200 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.161) Back Note:H1 = [Hf (reactants)] = 4 Hf [CH3·NH ·NH2(l)] + 5 Hf [N2O4(l)] H2 = [Hf (products)] = 4 Hf [CO2(g)] + 12 Hf [H2O(l)] + 9 Hf [N2(g)] Applying Hess’s law, H1 + H = H2 H = H2 - H1 = (4 Hf [CO2(g)] + 12 Hf [H2O(l)] + 9 Hf [N2(g)] – (3 Hf [CH3·NH ·NH2(l)] + 5 Hf [N2O4(l)] = [4 (-393.5) + 12 (-285.8) + 9 (0) – 4 (+53) – 5 (-20)] kJ mol-1 201 =- 5115.6 kJ mol-1 The standard enthalpy change of the reaction is –5115.6 kJ mol-1. 6.6 Calculations involving standard enthalpy changes of reactions (SB p.162) Given the following information, find the standard enthalpy change of formation of methane gas. C(graphite) + O2(g) CO2(g) Hc [C(graphite)] = -393.5 kJ mol-1 1 H2(g) + O2(g) H2O(l) Hc [H2(g)] = -285.8 kJ mol-1 2 CH4(g) + 2O2(g) CO2(g) + 2H2O(l) Hc [CH4(g)] = -20 kJ mol-1 Answer 202 6.6 Calculations involving standard enthalpy changes of reactions (SB p.162) Direct measurement of ΔHf [CH4(g)] is impossible because carbon(graphite) and hydrogen do not combine directly, and methane does not decompose directly to form carbon(graphite) and hydrogen. Since methane contain carbon and hydrogen only, they can be related to carbon dioxide and water by the combustion of methane and its constituent elements as shown in the diagram below. 203 6.6 Calculations involving standard enthalpy changes of reactions (SB p.163) Back Note: H1 = Hc [C(graphite)] H2 = 2 Hc [H2(g)] H3 = Hc [CH4(g)] Applying Hess’s law, Hf [CH4(g)] + H3 = H1 + H2 Hf [CH4(g)] = H1 + H2 - H3 = Hc [C(graphite)] + 2 Hc [H2(g)] - Hc [CH4(g)] = [-393.5 + 2 (-285.8) –(-890.4)] kJ mol-1 = -74.7 kJ mol-1 The standard enthalpy change of formation of methane gas is –74.7 kJ mol-1. 204 6.6 Calculations involving standard enthalpy changes of reactions (SB p.163) Given the following information, find the standard enthalpy change of formation of methanol. C(graphite) + O2(g) CO2(g) Hc [C(graphite)] = -393.5 kJ mol-1 1 H2(g) + O2(g) H2O(l) Hc [H2(g)] = -285.8 kJ mol-1 2 C2H5OH(l) + 3O2(g) 2CO2(g) + 3H2O(l) Hc [C2H5OH(l)] = -1371 kJ mol-1 Answer 205 6.6 Calculations involving standard enthalpy changes of reactions (SB p.163) 206 6.6 Calculations involving standard enthalpy changes of reactions (SB p.163) Back Note: H1 = 2 Hc [C(graphite)] H2 = 3 Hc [H2(g)] H3 = Hc [C2H5OH(l)] Applying Hess’s law, Hf [C2H5OH(l)] + H3 = H1 + H2 Hf [C2H5OH(l)] = H1 + H2 - H3 = 2 Hc [C(graphite)] + 3 Hc [H2(g)] - Hc [C2H5OH(l)] = [2 (-393.5) + 3 (-285.8) –(-1371)] kJ mol-1 = -273.4 kJ mol-1 The standard enthalpy change of formation of ethanol is –273.4 kJ mol-1. 207 6.6 Calculations involving standard enthalpy changes of reactions (SB p.164) (a) Find the standard enthalpy change of formation of butane gas (C4H10(g)). Given: Hc [C(graphite)] = -393.5 kJ mol-1 Hc [H2(g)] = -285.8 kJ mol-1 Hc [C4H10(g)] = -2877 kJ mol-1 208 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.164) Hf [C4H10(g)] = Hc [C(graphite)] 4 + Hc [H2(g)] 5 - Hc [C4H10(g)] = [(-393.5) 4 + (-285.8) 5 – (-2877)] kJ mol-1 =209-126 kJ mol-1 6.6 Calculations involving standard enthalpy changes of reactions (SB p.164) (b) Find the standard enthalpy change of the reaction: Br2(l) + C2H4(g) C2H4Br2(l) Given: Hf [C2H4(g)] = +52.3 kJ mol-1 Hf [C2H4Br2(l)] = -80.7 kJ mol-1 210 Answer 6.6 Calculations involving standard enthalpy changes of reactions (SB p.164) Back H = [Hf (products)] - [Hf (reactants)] = [-80.7 – (+52.3) – 0)] kJ mol-1 = -133 kJ mol-1 211 6.7 Entropy change (SB p.167) Back Predict whether the following changes or reactions involve an increase or a decrease in entropy. • Dissolving salt in water to form salt solution • Condensation of steam on a cold mirror • Complete combustion of carbon (a) Increase (b) Decrease (c) Increase • Complete combustion of carbon monoxide (d) Decrease • Oxidation of sulphur dioxide to sulphur trioxide (e) Decrease 212 Answer 6.8 Free energy change (SB p.170) Back In the process of changing of ice to water, at what temperature do you think G equals 0? G equals 0 means that neither the forward nor the reverse process is spontaneous. The system is therefore in equilibrium. Melting point of ice is 0 oC (273 K) at which the process of changing ice to water and the process of water turning to ice are at equilibrium. At 0 oC, G of the processes equals 0. 213 Answer 6.8 Free energy change (SB p.170) (a) At what temperatures is the following process spontaneous at 1 atmosphere? Water Steam (a) 100 oC (b) What are the two driving forces that determine the spontaneity of a process? (b) Enthalpy and entropy Answer 214 6.8 Free energy change (SB p.170) Back (c) State whether each of the following cases is spontaneous at all temperatures, not spontaneous at any temperature, spontaneous at high temperatures or spontaneous at low temperatures. (i) positive S and positive H (ii) positive S and negative H (iii) negative S and positive H (iv) negative S and negative H (i) Spontaneous at high temperatures (ii) Spontaneous at all temperatures (iii) Not spontaneous at any temperature (iv) Spontaneous at low temperatures 215 Answer 6.5 Hess’s law (SB p.153) Enthalpy change of formation of CaCO3(s) Ca(s) + C(graphite) + 3 O2 2 Hf [CaCO3(s)] CaCO3(s) H2 H1 CaO(s) + CO2(g) Hf [CaCO3(s)] = H1 + H2 = -1028.5 kJ mol-1 + (-178.0) kJ mol-1 = -1206.5 kJ mol-1 216 6.5 Hess’s law (SB p.153) Enthalpy change of hydration of MgSO4(s) MgSO4(s) + 7H2O(l) ΔH1 ΔH aq MgSO4·7H2O(s) aq ΔH2 Mg2+(aq) + SO42-(aq) + 7H2O(l) ΔH = enthalpy change of hydration of MgSO4(s) ΔH1 = molar enthalpy change of solution of anhydrous magnesium sulphate(VI) ΔH2 = molar enthalpy change of solution of magnesium sulphate(VI)-7-water ΔH = ΔH1 - ΔH2 217