Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
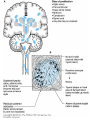
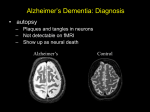
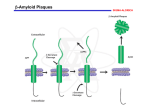
Demyelinating Diseases • Demyelination is a common degenerative change in the nervous system • secondary to neuronal or axonal injury, • But in the group of diseases known as the demyelinating diseases, demyelination is the primary pathologic process Multiple Sclerosis • The most common demyelinating disease. • Most common in the scandinavian countries, with a prevalence of 80:100,000 in norway. The incidence progressively declines as one moves south (10:100,000 in southern europe). • Rare in the tropics (1:100,000) and in asia, • The onset years from 20 to 40. • Sixty percent of patients are female. • (Caucasians more commonly affected than african-americans or native americans). • Increased familial incidence, • A 25% concordance in identical twins compared with 2–3% in fraternal twins, Etiology • Immunologically mediated demyelination, possibly acting via damage to oligodendroglial cells, which are consistently absent in lesions. • activation of macrophages and T lymphocytes • Increased immunoglobulin synthesis • In both blood and cerebrospinal fluid during the active phase of the disease. • Activated T lymphocytes are present in the lesions of multiple sclerosis Pathology • Presence in the white matter of plaques of demyelination. • These plaques are perivenular • Appear as irregular, • Well-demarcated, gray or translucent lesions • Diameter varying from 0.1 cm to several centimeters. • Multiple plaques, widely disseminated throughout the central nervous system, are common • • • • Any area of the brain can be affected. Optic nerves, Paraventricular regions, Brain stem, cerebellum, spinal cord, and deep cerebral white matter. Microscopically, • The plaques show demyelination • Tangled masses of preserved axons • Lymphocytic infiltration is present in areas of active and recent demyelination. • Macrophages with phagocytosed myelin. • Reactive astrocytic proliferation at the edges of the plaque. • Oligodendroglial cells are typically absent in the plaque. Clinical Features • Chronic disease with an extremely variable clinical course, • Episodic relapses and remissions over several years. • Mean survival is over 30 years after the onset of disease. • A minority rapid course to death within months, • Some appear to have only one or a few episodes Common manifestations are • Abnormalities in vision, cerebellar dysfunction, paresthesias, weakness, and spinal cord dysfunction. • The randomly disseminated nature of the lesions gives a characteristic clinical picture when multiple plaques are present. Degenerative Diseases Cerebrocortical Degenerations Alzheimer's Disease • Extremely common • Responsible for more than 50% of all cases of dementia • Characterized by progressive loss of neurons in the entire cerebral cortex. • The frontal lobe is involved preferentially. • Neuronal loss leads to dementia, which is the characteristic clinical presentation • Initially applied dementia under 65 years (presenile dementia), • After age 65 was called senile dementia. • The changes seen in most patients with senile dementia are identical to those of alzheimer's disease. • Alzheimer's disease occurs in 20% of persons over 80 years old. Etiology • Unknown. • Abnormalities of chromosomes 14, 19, or 21 have been identified in affected families, • Patients with down syndrome (trisomy 21) frequently develop lesions of alzheimer's disease in the third or fourth decade of life. Pathology • Grossly, there is atrophy of the cerebral cortex, • With thinning of the gyri and widening of the sulci • Affecting the frontal parietal and medial temporal lobes. • The cortical gray matter is greatly thinned and poorly demarcated. • The lateral ventricles show compensatory dilatation. Microscopically • Neuronal loss and disorganization of the cerebrocortical layers. • Presence of neurofibrillary tangles in the cytoplasm of affected neurons • These are complexly interwoven masses of paired helical filaments 10 nm in diameter consisting of various proteins, • Neuritic plaques, which are large (150 m) extracellular collections of degenerated cellular processes disposed around a central mass of -amyloid protein material • The degenerated neuritic material contains paired helical filaments identical to those found in neurofibrillary tangles in affected neurons. Neuritic plaques in cerebral cortex in Alzheimer's disease, showing cellular processes disposed around a central mass of -amyloid Clinical Features • • • • • • Over 50 years of age. Loss of higher cortical functions. The loss of ability to solve problems, Decreased agility of thought processes Mild emotional lability The dementia progresses inexorably over the next 5–10 years to an extent that the patient becomes unable to carry out daily activities. • There is no effective treatment Huntington's Disease(Huntington's chorea) • rare disease • inherited as an AUTOSOMAL DOMINANT TRAIT with complete penetrance but delayed appearance. • The abnormal gene is located on the terminal segment of 4p. • DNA probes are now available to detect the abnormal gene in affected families before symptoms develop. • This provides crucial information for genetic counseling • Atrophy and loss of neurons of the caudate nucleus and putamen, • Associated with variable cerebrocortical atrophy, particularly in the frontal lobe. • There is a marked decrease in synthesis of the neurotransmitter -aminobutyric acid in the basal ganglia. • Though inherited, the disease has its onset in adult life, • Usually between 20 and 50 years of age. • It is characterized by dementia, due to cerebral involvement, and • Choreiform involuntary movements, due to involvement of the basal ganglia. • The disease is slowly but inexorably progressive, leading to death in 10–20 years.