Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
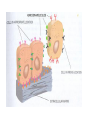
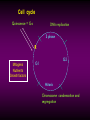
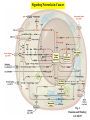
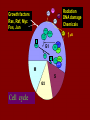
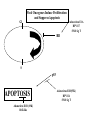
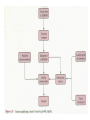
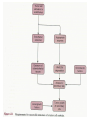
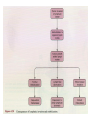
STUDY OF CANCER & Immune System LS601 Professor Swapan K. Ghosh [email protected] 237-2416 Lectures From April 4-15,2003 Fact Sheet: In 1996 554,740 deaths due to cancer in US alone. Over 8 million cancer patient in US are alive 5 million of those were diagnosed 5 years ago. One of approx. 3 will experience cancer “CANCER IS A CRIMINAL-IT DOES NOT FOLLOW RULES” Dr. S. Otani CANCER: The term refers to 100 forms of the disease. Almost every tissue can get transformed into malignancy. Caused by : 1. Uncontrolled growth by a cell that constitutes the tissue; 2. The resulting impact on normal functioning of body organs and disruption. 3. The ability of the aberrant cell to migrate to a distant site and invade neighboring tissues, a phenomenon called METASTASIS . Overview of cancer and carcinogenesis • Biology of cancer: – Microevolutionary process • Different stages of cellular transformation • Basic properties in cancer cells • Etiology: aberrant natural selection • Molecular genetics of cancer: Oncogenes, Tumor suppressor genes Microevolutionary processes leading to cancer • Animal bodies represent a society or ecosystem – Individual members are cells that reproduce and organize into assemblies or tissues – Involves cell births, deaths, territorial boundary, population sizes, species propagation – BUT virtually no competition for survival and all cells collaborate and sacrifice to produce germ cells ensuring propagation (All somatic cells die leaving no progeny) 1. Natural Selection in cells may disturb collaboration & predispose to cancer 2. Mutation, Competition and natural selection among somatic cells create a microevolutionary environment promoting growth of selfish aberrant cells or cancer SPECIFIC TOPICS General characteristics, classification, and nomenclature of tumors Properties: How cancer arises, How cancer spread Grading and staging Immunity and cancer Therapy: surgery, radiation, chemotherapy and biotherapy (immunotherapy) Classification: Carcinoma: cancer of epithelial tissues Adenocarcinoma: cancer of glandular tissue; spread through lymphatics Sarcoma: cancer of stromal or mesenchymal layers of organs; spread via blood. Carcinosarcoma: mixtures of cancer cells from both epithelia and mesenchma. Teratoma: cancer of stem cells. Undifferentiated neoplasms: poorly undifferentiated. Terms and Definitions Neoplasia : New aggressive growth of cells and tissues putting pressure on neighboring tissues (Causing abnormal swelling or tumor) or invading neighboring tissues (cancer) Hyperplasia: Means too much cell proliferation or mitosis. This is abnormal but not cancer Dysplasia: A cell is not only proliferating excessively, but attains abnormal and orientation; Pre-cancerous More TERMS: Metaplasia: conversion of one cell type into another, such as, stratified oesophagus or lung tissues (due to acidity or cigarette).The process is reversible and is not cancer, but may lead to cancer. Metastasis: Spreading to distant sites. - First site where cancer is detected is called primary site and the second site, secondary site. -Small clumps of cancer cells (emboli) Spread by migration through blood (called blood-borne or hematogenous) or through lymphatics (lymphogenous). -Cancer cells spread because they lose their molecular address where to go. More Jargons! Anaplasia: Primitive undifferentiated state of cell growth. Aplasia: A loss of normal appearance and disorganizations of tissues. In situ cancer: abnormal growth at a particular site but no invasion of neighboring tissues. Benign and fibrous. Invasive cancer: Lethal and malignant as neighboring tissues are invaded. Characteristics of cancer cells 1. Infiltration and destruction of surrounding tissues. 2. Loss of contact inhibition of growth. Anchorage independent and aberrant chromosome numbers or aneuplody 3. Variation in shapes and sizes based on degree of differentiation. 4. Uncontrolled mitosis or cell proliferation or growth rate. Less dependent on growth factors 5. Often migration to distant sites and loss of similarity with parent tissues. Cancer classification • Sporadic cancer: cancer without a family history; nonhereditary and not affecting off-springs Mutations not present in the germline cells. Colon cancer mostly sporadic • Hereditary cancer: – Mutations are present in the germline cells and predispose to inheritance towards developing cancer (familial). Breast cancer is an example Most Cancers originate from a single abnormal cell (Clonal origin) • Cancer is essentially a genetic disease but not, in most cases, an inherited disease • Cancer is nearly always of clonal origin • Multiple mutations (at least TWO-HIT theory as proposed by Knudson) in cancer cells • Same genes are often altered by carcinogens, radiation, viruses undergo translocation, amplification or deletion Cancer cells differently from normal cells Experimental evidence of cancer clonality •If cancer originates from one mutated cell, it could be traced by looking at fingerprints of genes and/orproducts •Tracking the identity of a marker X-chromosome that is inactivated in cancer cells in females •Studying chronic myelogeneous lymphomas (CML) and tracking Ph chromosome as markers •Studying Myelomas in which Myc oncogene has translocated only to a specific Ig gene X-Chromosome inactivation in Females •Only one of the two X chromosomes inherited are inactivated in a cell. • If cancer is monoclonal origin, it would clearly have only one X-chromosome inactivated. •No mixed inactivation be seen in any given tumor How To Demonstrate Clonal Origin of Cancer Cells MYC GENE TANSLOCATION IN MYELOMAS Karyotype of a breast cancer cell depicting translocations (DIFFERENT COLORS) Abnormal cells in Pap smear What causes cancer Various factors Contribute to the cause of cancer. Carcinogens, that is, cancer producing agents can be of physical, chemical, or of biological nature. •Physical: Radioactivity, UV radiation, X-rays. •Chemicals: from cigarette smokes, and shoots of chimney or coal: hydrocarbon (benzpyrene), DMBA, aromatic amine (in many synthetic dyes). •Dietary factors: such as saturated fats, food additives, lack of fruits and vegetables (antioxidants), alcohol, excess meat products Mutations by carcinogens What causes cancer •Biological factors: hormones and viruses: estrogen and pituitary hormones promotes breast tumor. •Oncogenic viruses: leukemia viruses (Rous sarcoma virus) , Epstein-Barr virus (Burkitt’s lymphoma), Papilloma virus, Mammary tumor virus, Simian virus How Cancer Arises 1. Cancer cells violate the civic rules that govern normal cells by not responding to go-signals for proliferation and stop-signals for reproduction. 2. Cancer cells descend from a common ancestral cell: clonal origin. But at some point one of the off-springs mutate that becomes worse with more mutation, and finally the accumulated mutated cells disobey all civic controls of normal cells in a tissue, becoming invasive and malignant. 3. Since mutations occur at the gene level, that is, DNA molecules that reside in the nuclei of the cells, most human cancer can be traced there. Proto-oncogenes from neighboring cells produce growth factors that encourage cell growth during cell cycle by producing growthstimulatory signals. Mutation in these genes may cause cells divide without any signal from outside. One example is mutated ras gene. A quarter of all human tumors have mutated ras gene. Similarly myc gene family if abnormal causes leukemia, lymphomas. Receptors on the recipient cells that bind proto-oncogene growth factors may also mutate and stimulate cell growth.Thus, in breast cancer, Erb-B2 receptors behave abnormally. Tumor suppressor genes that control unrestricted growth of cells and inhibit cell growth. If they stop working, cancer cells grow wild and uninterrupted. Example, pRB, P53 (tumor-suppressor gene protein), TGF-beta (inhibits cell growth) Cell cycle clock malfunction : Cyclin protein binds to cyclin kinase (CDK) and releases the braking action of p53 on cell proliferation from G1 to s phase. In cancer p53 is inactive, so cells keep growing. Apoptosis or programmed cell death. Normally, if a cell is abnormal p53 gene product will promote suicide of the cell thus avoiding cancer. If p53 does not function the abnormal gets to live and be cancerous. Cancer cells may Overcome p53’s grip on cell cycle by producing excess of Bcl-2 protein that counteracts the action of p53 All these make cancer cells immortal and cancerous. How Cancers Spread Cancer cells are malignant cells. What is malignancy? It means invasiveness. If a tumor does not disturb or infiltrate into neighboring cells, they are BENIGN tumors. But if they invade, they become MALIGNANT and cancerous. AND IF They spread they become metastatic cancer. Invasion of neighboring cells and tissues: 1. Cancer cells lose their area code. It means that they do not have glue or adhesion molecules with which to attach to specific cells and form organs. 2. Cancer cells invade adjoining tissues by releasing enzymes, called metalloproteinases, that dissolve basement membranes and other extracellular matrix. Once detached from where the cancer cell belongs, and destroying the surrounding matrix, it goes into blood vessels or lymphatics, gets carried to distant sites, and establish metastatis. Invasion and metastasis Cancer Genetic Initiation (Oncogenes/ tr suppressor genes) Epigenetic Promotion Progression (clonal expansion) Mitogenesis Immune surveillance Angiogenesis TUMOR SUPPRESSOR GENES AND CANCER Mutation 2 Mutation 1 Normal X Heterozygous Other: Gene Methylation Expression Levels X X X Loss of Heterozygosity Cell cycle Quiescence = Go DNA replication S phase R Mitogens Nutrients Growth factors G2 G1 Mitosis Chromosome condensation and segregation General Cell Cycle Facts Cell cycle is a fundamental cellular process Wonderful convergence of cell biology, biochemistry and genetics Principles are conserved in xenopus, yeast and mammals G1 S G2 M Go -restriction point; start -DNA replication -"rest" phase -mitosis -quiescence Can be variable lengths. Mammalian cells in culture: 14hr. Liver cells: 1 yr (except under regeneration) Kinase Activity A Kinase Machine Cyclin D1 CDK 4 G1 Cyclin E CDK 2 Cyclin A CDK 2 S Cyclin B CDC 2 G2/M Cell cycle clock malfunction : Cyclin protein binds to cyclin kinase (CDK) and releases the braking action of p53 on cell proliferation from G1 to s phase. In cancer p53 is inactive, so cells keep growing. Apoptosis or programmed cell death. Normally, if a cell is abnormal p53 gene product will promote suicide of the cell thus avoiding cancer. If p53 does not function the abnormal gets to live and be cancerous. Cancer cells may Overcome p53’s grip on cell cycle by producing excess of Bcl-2 protein that counteracts the action of p53 All these make cancer cells immortal and cancerous. Signaling Networks in Cancer Fig. 2 Hanahan and Weinberg Cell 100:57 Oncogenes Gene encoding proteins that is essential for the initiation, promotion and progression of the malignant state >100 different oncogenes discovered as the transforming genes of RNA tumour viruses Proto-oncogene counterpart of viral-oncogene - normal cellular genes which encodes proteins to regulate cellular response to external stimuli that controls cellular proliferation and differentiation. - activated by mutation - nomenclature Classification of oncogenes 1. Growth factors - sis, ist 2. Growth factor receptors (RTK) - erb B2, fms 3. Non receptor tyrosine kinases - abl, src 4. GTP binding - ras 5. DNA damage repair - ATM, MSH2, B cl2 6. Serine/ threonine kinases 7. Nuclear binding - Myc, fos, jun 8. Misc - cell surface APC/ DCC Tumor suppressor gene and Oncogenes exert opposite effects Proto=oncogenes and Suppressor genes activated by DNA damage How Rb controls cell cycle Conversion of a proto-oncogene to an oncogene Proto-Oncogene products A model for p53 function RB gene mutations lead to cancer Mechanisms of Proto-oncogene activation 1. Mutations: * Point mutations/deletions/insertions - most characterized in human trs, frequent in Ras * Causes constitutive activation of the signal transducing function of Ras protein 2. Gene amplification Results from several rounds of unscheduled DNA synthesis occurring during a single cell cycle - HSR, DM Her2 Breast cacinoma, n-myc neuroblastoma 3. Chromosomal re-arrangement * chromosomal translocations; inversions * Gene activation -transcriptional activation of Proto-oncogenes eg. follicular lymphoma t (14:18) Bcl2 Burkitts lymphoma t (8:14) c-myc * Gene fusion: codes for chimeric protein eg. CML t (9:22) BCR/Abl. APML t(15:17) PML/Rar Ras - super gene family – a group of G-proteins – contains >50 proteins – H-ras, K-ras, N-ras – N-ras mutation most common alteration seen in cancer – Prenylation - Farneysl transferase Ras activation in Leukemia CMML - 32-65%, AML - 25-44%, ALL 6-18% * Other mechanisms of activation Y SOS G GRB2 adaptor mol. S P G S R Raf (MAPKKK) GTP GDP MAPKK MAPK Ras - G protein system Fos/ Jun activation Cyclin D1 Ras GAP Tumour suppressor genes: * recessive genotype - exception p27kip1 * Controls cell proliferation * >20 tumor suppressor genes * Inherited cancers: Colon cancer Retinoblastoma, Wilm’s tr, breast cancer Neurofibromatosis,Li-Fraumeni syndrome, Xeroderma pigmentosum p19 p18 Growth factors Ras, Raf, Myc Fos, Jun p27 p15 Radiation DNA damage Chemicals Ckd 2,4,6 CD 1,2,3 E2F p107 G1 E2F p21 PCNA CE Cdk2 Rb CA P p107 M S G2 Cell cycle Cdk2 p53 DNA damage MDM2 p53 P21 Bax/Bcl2 Waf1/ Cip1 Apoptosis G1/S Cyclin D/ cdk R Rb P pRB + E2F S G1 E2F G2 Cell cycle damage and control M Angiogenesis Proangiogenic - Angiogenic growth factors VEGF, FGF, TNF, HGF, IGF,TGF, Proliferin Anti angiogenetic - Thrombospondin -1, 2 Angiostatin Endostatin Platelet factor 4 Apoptosis pathways Caspase 9 Apaf 1 Apoptotic signal Caspase 3 Cyt C Bcl2 AIF Apoptosis .. Cyt C N nucleus Mitochondria Bax p53 G1 Viral Oncogenes Induce Proliferation and Suppress Apoptosis Adenovirus E1A HPV E7 SV40 Lg T RB S p53 APOPTOSIS Adenovirus E1B (19K) Bcl2-like Adenovirus E1B(55K) HPV E6 SV40 Lg T pRB Pathway Mitogenic Stimuli (e.g. GF, Ras) RB E2F D-Cyclin CDK 4/6 E2F P16 Ink4a X DNA Pol Cyclin E, p19 DHFR, MYB PPP RB Tumor Suppressor Genes RB, p16 Oncogenes Cyclin D1 From Sharpless and DePinho (1999) Current Opinions in Genetics and Dev. 9:22 SENSOR of DNA DAMAGE p53 increased levels phosphorylation conformational change oligomerization Transcriptional Activation Cell Cycle Repair Apoptosis p21 mdm2 GADD45 BAX Figure 1 Levine (1997) Cell 88:325 P53 and RB Crosstalk p53 Ink 4 p19 p16 Bax E2F1 mdm2 CyclinD CDK4 RB BCL2 Caspases E2F1 Apoptosis S-Phase Apoptosis p19 Similarity of Cell Cycle and Apoptotic Machinery Apoptosis Cell Cycle A protease machine A kinase machine Caspases CDKs (Cysteine protease which cleaves after ASPartate ) (Cyclin-dependent Kinase) Inactive pro-enzyme Activated by proteolytic processing cascade Blocked by IAP (inhibitors of apoptosis) Inactive Activated by kinases, phosphatases Blocked by CDKI Evolutionarily Conserved Evolutionarily Conserved Ways to Score Apoptosis Chromatin and Nuclear Condensation Morphology on Hoechst dye immunofluorescence Normal: round nuclei. Apoptotic: irregular and condensed DNA fragmentation (most often employed for apoptosis) Appearance of a DNA ladder (due to intranucleosomal degradation) Sub G1 population in FACS TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) Membrane changes (early events) Annexin binding binds phosphatidylserine generally on the inside of the plasma membrane, but flips Cleavage of PARP by Caspases Intrinsic Apoptosis Pathway Figure 3 Wang, X. (2001) GenesDev. 15:2922 Bcl2 gene Family bcl-2 was identified as a gene that was translocated in B-cell lymphomas. --Unlike most oncogenes, bcl 2 extended cell survival, rather than promote G1. -Withdrawal of survival factors (e.g. IL-3) leads to apoptosis, but bcl-2 protects. Bcl2 Family of Apoptotic Regulators Both pro- and anti-apoptotic members. Anti-apoptotic: Bcl2, BCL-X-L Pro-apoptotic: BAX , BAD All have bcl-2 homology region (BH1-3) All interact with each other. e.g. BAX/bcl2, bcl-x ratio is a determinant of death All affect mitochondrial membrane permeability. Anti-apoptotic members block cytochrome c and SMAC release. G1 G0 Jun, FOS p53 M Cyclin D CDK4 p16 Ink 4a G2 p21 Cyclin E CDK2 E2F RB Cyclin A CDK2 DP PP PP E2F DP (inactive E2F) PPP S RB E2F DP (active E2F) Tissue Differentiation Intrinsic and Extrinsic Death Pathway Extrinsic Intrinsic Figure 2 Johnstone et al (2002) Cell 108:153 Treatment approaches in Oncology Surgery Radiotherapy Chemotherapy Biotherapy (experimental): Immunotherapy (magic bullet) Gene therapy Bone marrow transplantation How do you select treatment modality? After pathological determination of the tumor type, and TUMOR STAGING What is tumor staging? Categorizing malignant tumors relative to their potential for invasiveness and metastatic capability. Stage I = tumor at the primary site; Stage II & III= moderate spread and invasion; Stage IV= extensive spread and invasion (poorly differentiated, pleomorphic, visible mitoses; least chance of survival). Another way of tumor grading: TNM system where T means how big the tumor is at the primary site; N means enlargement of regional lymph node; M means extent of metastasis. hat does ToNoMo mean? Surgery: good for benign tumor, less for malignant tumor depending on the stage of the tumor. For invasive tumor removal of lymph node may be necessary. Raises concern for iatrogenic spread. Radiotherapy: Destructive dose of ionizing radiation from cobalt-60 may kill tumors. Since it raises temperature, it causes burn in skin other non tumor areas. Not all tumors are radiosensitive. Chemotherapy: Interferes with metabolism or mitosis of tumors. Not always work. Harmful for normal tissues too. Often tumors develop resistance. (MDR expression) Combination therapy: Radiation + Chemo. As well as a combination of various drugs. Immunotherapy : potentially the best treatment. Immunotherapies: biological response modifiers (Cytokines), anti-tumor antibody, anti-tumor antibodytoxin conjugates (magic bullet), LAK (lymphokineactivated killer) cells, Adjuvants. Gene Therapy: Inserting missing genes into cancer cells Inserting anti-oncogene DNA Inserting tumor-supressor genes such as p53, pRB DNA vaccine Figure 3. The cell cycle clock and cancer. The cell cycle clock-composed of an assembly of interacting proteins in the nucleus-normally integrates messages from the stimulatory and inhibitory pathways and, if the stimulatory messages win out, programs a cell's advance through its cycle of growth and division. Progression through the four stages of the cell cycle (a) is driven by rising levels of proteins called cyclins: the D type, followed by E, A and B. A crucial step in the cycle occurs late in G1 at the restriction point (R), when the cell decides whether to commit itself to completing the cycle. For the cell to pass through R and enter S, a molecular "switch" (b) must be flipped from "off" to "on". As levels of cyclin D cyclin E rise, these proteins combine with and activate cyclin-dependent kinases (1). The kinases grab phosphate groups (2) from molecules of ATP (adenosine triphosphate) and transfer them to protein pRB, the master brake. When pRB lacks phosphates, it actively blocks cycling (and keeps the switch in the "off" position) by sequestering other proteins termed transcription factors. But after the cyclin-kinase complexes add enough phosphates to pRB, the brake stops working (3); it releases the factors, freeing them to act on genes. The liberated factors then spur production of various proteins required for progression through the cell cycle. In c, the switch is placed in the larger context of the many molecular interactions that regulate the cell cycle. Flipping the switch to "on" can be seen above the R point. Over-activity of the stimulatory proteins cyclin D, cyclin E and CDK4 have been implicated in certain human cancers. Inactivation of various inhibitory proteins has also been documented. The affected proteins include p53, pRB, p16 and p15. Mitochondria in apoptotic signaling Clinical Applications 1. Diagnosis - oncogenes c-myc in Burkitts Bcr/Abl CML 2. Detection of MRD 3. Prognosis Bcr/Abl CML n-myc neuroblastoma Her 2 breast cancer 4. Predictive oncology MEN Type II, Li-Fraumeni syndrome Retinoblastoma, Wilms tr, Breast ca.