Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
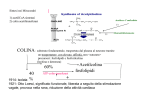
ACh was first isolated around 1914; its functional significance was first established in about 1921 by Otto Loewi, a German physiologist and later (1936) Nobel laureate. Loewi demonstrated that ACh is the substance liberated when the vagus nerve is stimulated, causing slowing of the heartbeat. Subsequently he and others showed that ACh is also liberated as a transmitter at the motor end plate of striated (voluntary) muscles of vertebrates, and it has since been identified as a transmitter at many neural synapses and in many invertebrate systems as well Sintesi nei Mitocondri 1) acetilCoA sintetasi 2) colin acetiltransferasi Acetilseco 3’emilcolinio (-) (-) 2benzoiletiltetramonio Composti organici del merucurio COLINA: 40% substrato fondamentale, trasportata dal plasma al neurone tramite un trasportatore con elevata affinità, non <saturato> precursori: fosfolipidi e fosforilcolina (lecitina e demenza) Acetilcolina 60% ATP-colin-transferasi + fosfolipidi Storage and release ACh in cholinergic nerve fibers is taken up into synaptic vesicles by an uptake process that is inhibited by the drug vesamicol. In the presence of vesamicol, cholinergic fibers soon have no ACh stored in vesicles for release. Transmission fails although other functions of the fiber are still intact Vesicular release depends on depolarization of the nerve terminal and the influx of calcium ion. At the motor end-plate in the neuromuscular junction this results in a relatively massive release of ACh (hundreds of vesicles and thousands of ACh molecules per vesicle) and an end-plate potential that normally results in depolarization of the muscle cell and contraction. The release of ACh at various cholinergic junctions can be blocked by certain toxins, most notably those produced by Clostridium species. Botulinum toxin A, from Clostridium botulinum binds to cholinergic nerve terminals and is internalized. Once internalized it acts on the vesicle release process and prevents exocytosis. All junctional release of ACh is inhibited by such toxins. In patients poisoned by Clostridium botulinum the immediate clinical problem is flaccid paralysis and respiratory failure. Botulino: blocco rilascio Vedova nera: aumenta rilascio Curaro: blocca i recettori post-sinaptici ACETILCOLIN ESTERASI Assicura l’efficienza della neurotrasmissione colinergica Ciclo del messaggio chimico : 2 msec nella trasmissione neuromuscolare 1 msec muscolo liscio Sede: dendriti e nel pericarion dei neuroni, collocato nello spazio sinaptico legato ad una rete di collageno che forma la lamina basale che riempie lo spazio tra neurone e cellula muscolare striata Acetylcholine (ACh) is terminated by hydrolysis, which is greatly accelerated by one or more of the cholinesterase enzymes: 1) Acetylcholinesterase (AChE) is present in high concentration in cholinergic synapses (SNC, muscolo sch . 2) Butyrylcholinesterase, also known as pseudocholinesterase is important for hydrolyzing ACh in the circulation. (Fegato intestino cuore e polmoni) It is important to recognize that the neurotransmitter actions of acetylcholine are terminated by a chemical reaction that forms two products (choline and acetate) which are essentially inactive. Diffusion of ACh from the synaptic region plays a minor role because AChE is so active. AChE inhibitors, also designated AChEIs, include echothiophate, edrophonium, neostigmine, physostigmine. Other AChEIs include various so-called nerve gas agents such as sarin and soman. Ach E: inibizione substrato, BchE attiva solo ad alte concentrazioni di substrato (rappresenta una riserva di AchE se qs è poca o assente, come durante differenziamento e sviluppo cell. Actions of acetylcholine Acetylcholine (ACh) has diverse actions on a number of cell types mediated by two major classes of receptors: 1) Nicotinic receptors are ligand-gated ion channels. 2) Muscarinic receptors are part of the transmembrane, G protein coupled receptor family. \ NICOTINIC RECEPTORS 1) nicotinic muscle (Nm): neuromuscular junction of skeletal muscle; 2) nicotinic neuronal (Nn): autonomic ganglia and other parts of the nervous system When ACh or other agonists occupy the receptor site on the external surface of the cell membrane there is a conformational change in the ion channel and an increase in conductance to the ion(s) for which that channel is selective. Thus, when Nm receptors are activated, there is an influx of cations through the ion channel and depolarization of the motor end plate. In short, nicotinic receptors rather directly transduce the ACh external messenger into an action on the cell. The acetylcholine receptor is a pentaramic protein consisting of five subunits (2 alpha units, one beta unit, one gamma unit, and one delta unit); each subunit encoded by a seperate gene. For all five subunits to assemble correctly the gene expression must be precisely coordinated. The five subunits are arranged in a barrel-like configuration around a central ion pore. Acetylcholine binds to the alpha subunit, which consists of 457 amino acids. The main binding site for acetylcholine is on the alpha subunit within a pocket of the external part of the peptide chain. Intracellular ions are collected within the folds of the receptor and attracted to charged residues within the walls of the folds. Residues are located at the ends of the pores to help determine the ionic selectivity of the channel: oppositely charged residues attract, therefore the negative receptors of an acetylcholine receptor attract cations. Acetylcholine reacts with the residues to form weak bonds which cause an alosteric change in the subunit configurations and allows ions to enter the channel. The channel is nonselective between cations, producing an inward flow of positive charges. These positive charges initiate the action potential which causes the muscle to contract. Nicotinic Receptors MUSCARINIC RECEPTORs Transduction of the ACh message is more complex in the muscarinic family of receptors. And the family of muscarinic receptors is more complex than the nicotinic family. There are at least 5 muscarinic receptor subtypes expressed in humans. For most purposes it is sufficient to concentrate on M1, M2 and M3 receptors. M1 p PKC 1) M1 receptors : autonomic ganglia central nervous system. 2) M2 receptors : > the supraventricular parts of heart the heart. 3) M3 receptors, smooth muscles and glands, endothelial cells in the vasculature. Correnti inibitrici K M2 legati a Gi inibiscono la del adenolato ciclasi e aprono i canali K The bottom line is that M1 and M3 receptors generally mediate excitatory responses in effector cells. Thus, M1 receptors promote depolarization of postganglionic autonomic nerves, and M3 receptors mediate contraction of all smooth muscles (an apparent exception to be noted below) and increased secretion in glands. It is useful to remember that excess ACh levels in the body (for example caused by inhibition of AChE) are associated with GI cramping, salivation, lacrimation, urination, etc. Muscarinic Receptors ACETYLCHOLINE RECEPTORS: Disorders * Muscle * Myasthenia Gravis * Autoimmune: IgG vs a1 subunit * Hereditary * Subunits: a & b * Subunit: e * Neuronal * Immune neuropathies: Isaac's; Subacute autonomic * IgG antibody vs a3 subunit * Paraneoplastic syndrome: Associated with small cell lung carcinoma * Epilepsy * Benign neonatal & Nocturnal frontal lobe, Type 1 l Neural nicotinic, a4 subunit ; Chromosome 20q13.2-q13.3; Dominant * Nocturnal frontal lobe, Type 3 l Neural nicotinic, b2 subunit (CHRNB2) ; Chromosome 1p21; Dominant * Schizophrenia: Attention disorder * Lack of inhibition of P50 response to auditory stimulus * Linked to dinucleotide polymorphism at 15q13-q14: Site of a-7-nicotinic receptor * Mouse knockouts * Lethal: e-AChR subunit loss * CNS neuronal loss with subunit knockout * Neural nicotinic, b2 subunit of AChR (CHRNB2) * Defects localized in CA1 and CA3 fields in hippocampus & neocortex * a7 subunit: Minimal phenotype * a9 subunit: Altered innervation of cochlear hair cells * Autonomic dysfunction * Knockouts of neural nicotinic AChR subunits * a3 : Bladder enlargement; Dilated, unresponsive pupils * b2 * Nicotine-elicited anti-nociception: Reduced * Neurons in hippocampus & neocortex: Reduced * * a4 Nicotine-elicited anti-nociception: Reduced * * * * * * * * Muscarinic IgG vs M3-muscarinic AChRs: Occur in both 1° & 2° Sjögren's Toxins Nicotinic agonists: Nicotine; Anatoxin A Nicotinic antagonists Peptides: a-snake toxins; a-conotoxins Other: d-tubocurarine; Histrionicotoxin; Lophotoxin; Epibatidine Muscarinic agonists: Muscarine; Arecoline; Pilocarpine; Green mamba snake Synaptic and Post-synaptic molecules at the NMJ MYASTHENIC & NEUROMUSCULAR JUNCTION (NMJ) DISORDERS BASIC CONCEPTS Acetylcholine receptors (AChRs) AChR structure AChR subunit mutations: a; b; e; d Neuromuscular junction (NMJ) Presynaptic Postsynaptic ACQUIRED NMJ DISORDERS Botulism Myasthenia gravis Autoimmune myasthenia gravis Childhood MG Drug-induced MG Neonatal: Transient MG Ocular Anti-AChR-Antibody-Negative Thymoma Domestic animals Myasthenic syndrome (Lambert-Eaton) Snake venom toxins ------------------------------------------------------------------------ CONGENITAL & FAMILIAL NMJ DISORDERS2 General features AChRs: Kinetic abnormalities Presynaptic defects Congenital MG + Episodic apnea (Familial infantile): ChAT; 10q11 Paucity of synaptic vesicles & Reduced quantal release Congenital Lambert-Eaton-like Episodic ataxia 2: CACNA1A; 19p13 Synaptic defects Acetylcholinesterase (AChE) deficiency at NMJs: ColQ; 3p25 Postsynaptic defects: AChR disorders Kinetic abnormalities in AChR function Reduced Numbers of AChRs at NMJs Increased Response to ACh: Slow AChR channel syndromes Delayed channel closure: AChR mutations Repeated channel reopenings: AChR mutations Reduced Response to ACh Fast-channel syndrome: Mode-switching kinetics D; AChR e subunit Fast channel syndrome: Gating abnormality; AChR a or e subunit Fast channel syndrome: Arthrogryposis; AChR d subunit Also see: e subunit disorders Normal numbers of AChRs at NMJs: Reduced Response to ACh Fast-channel syndrome: Low ACh-affinity of AChR; AChR e subunit Fast-channel syndrome: Reduced channel openings; AChR a subunit High conductance & Fast closure of AChRs Increased Numbers of AChRs at NMJs Slow AChR channel syndrome: AChR subunit bL262M No kinetic abnormalities in AChR function Reduced Numbers of AChRs at NMJs AChR mutations Usually: e subunit: 17 Rarely: a (2q24), b (17p12), d subunit (2q33) Rapsyn: 11p11 Other hereditary MG syndromes Benign congenital MG & Facial malformations Congenital MG: Other Familial immune Limb-girdle MG: Familial Plectin deficiency: Plectin; 8q24 The muscular weakness and fatigability associated with myasthenia gravis are caused by an autoimmune attack on the acetylcholine receptor at the neuromuscular junction. Antibodies have been shown to decrease the usefulness of acetylcholine receptors through accelerated endocytosis and blockade of the receptor. Endocytosis is the process of extracellular substances being incorporated into the cell by vesicles forming inward through budding of the plasma membrane. Researchers have been able to demonstrate the effect of antibodies on acetylcholine receptor degradation by using radioactively labeled alpha bungaroo toxin, a snake poison, to follow the rate of degradation. Antibodies from patients with MG cause a two to three fold increase in the rate of degradation of acetylcholine receptors. The myasthenic antibodies cause a cross linking between the acetylcholine receptors, causing the linked receptors to be drawn together into clusters and rapidly endocytosed. In myasthenic patients the neuromuscular junction has decreased numbers of acetylcholine receptors, a wider synaptic cleft, and simplified synaptic folds. These changes account for the clinical features of myasthenia gravis. Decreased numbers of acetylcholine receptors result in fewer interactions between acetylcholine and it's receptors, leading to decreased activation of action potentials. When the transmission of action potentials decreases, the power of the muscle's contraction is reduced, causing weakness. During repeated nerve stimulation the amount of acetylcholine normally declines, or runs down. In myasthenia gravis, this run down occurs more rapidly due to a decrease of receptors in myasthenic junctions, causing muscular fatigability. The wider synaptic cleft and simplified synaptic folds also work to decrease the number of interactions between acetylcholine and acetylcholine receptors.