Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
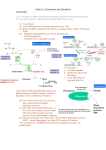
Hemostasis and Thrombosis Vic Vernenkar, D.O. St. Barnabas Hospital Dept. of Surgery Normal Hemostasis • A well regulated process • Maintains blood in a fluid, clot free state in normal vessels • Induces the rapid formation of a localized hemostatic plug at the site of vascular injury Thrombosis • Pathological state • Inappropriate activation of the normal hemostatic process – within the non-interrupted vascular system. • Thrombus (blood clots) formation – Blocks blood flow to vital areas Normal sequence of Hemostasis (4 steps) • 1. Arteriolar vasoconstriction (transient) – Reflex neurogenic mechanisms – Bleeding would resume after vasoconstriction if it weren’t for the activation of platelets or coagulation systems • 2. Exposure of subendothelial ECM when there is endothelial injury – ECM, especially collagen, is highly thrombogenic – Platelets adhere and become activated • Change in shape • Release of secretory products – Aggregation of platelets forms hemostatic plug – This is primary hemostasis First two steps of normal hemostasis Normal hemostasis continued • 3. Tissue factor released at the site of injury (by endothelial cells) – Works with secreted platelet factors – Activates coagulation cascade • A series of proteins where thrombin is activated • Induces further platelet recruitment and granule release – Ends in fibrin deposition – Called secondary hemostasis Normal hemostasis continued • 4. Formation of permanent plug – Prevents further hemorrhage – Polymerized fibrin and platelet aggregation – Counter regulatory mechanisms (t-PA) limit the plug to the site of the injury Steps 3 and 4 The Main Players in Hemostasis • Endothelial cells • Platelets • Coagulation cascade Endothelial Cells • Produce vWF (vonWillebrand factor) – A product of normal endothelium – found in the plasma – essential for platelet binding to collagen and other surfaces • Secrete Tissue factor – induced by cytokines (TNF, IL-1) – activates the extrinsic clotting pathway Endothelial Cells have Prothrombotic Effect • Via vWF and tissue factor • factors that depress fibrinolysis • factors needed for the clot are not destroyed before clot forms Collagen is highly thrombogenic For hemostasis For inflammation For wound healing Endothelial cells have Anti-thrombotic properties, too • Antiplatelet effects – Intact cells are barrier to subendothelial ECM – PGI-2 and NO prevent platelets from adhering • Fibrinolytic properties – Tissue type plasminogen activator (t-PA) • promotes activity to clear fibrin deposits from endothelial surfaces • Anticoagulant properties – Membrane associated molecules • Heparin like molecules and thrombomodulin – Inactivate thrombin and several coagulation factors Factors that favor or inhibit thrombosis SOooo….. • Endothelial cells modulate the balance of hemostasis • Endothelial injury is the dominant influence that leads to thrombosis Platelets • Express glycoprotein receptors on membranes. Gp Ib,IIb/IIIa • Have three types of granules – Alpha granules • Fibrinogen, fibronectin, factor V and VIII, PDGF, TGFb – Dense bodies or delta granules • ATP/ADP, ionized calcium, histamine, serotonin, epinephrine – Lysosomal granules Platelets Hyalomere and granulomere Platelets continued • Upon encountering the ECM, platelets undergo three general reactions: 1. Adhesion and shape change mediated by vWF and glycoprotein Ib 2. Secretion (release reaction) – calcium required in coagulation cascade – ADP as mediator of platelet aggregation – Surface expression of phospholipid complex • Binding site for calcium ions and coagulation factors Platelets continued • 3. Aggregation – ADP and TXA2 (vasoconstrictor thromboxane A2) are the stimuli for the formation of the primary hemostatic plug • Aspirin inhibits synthesis of TXA2 – Fused mass of platelets • Created by coagulation cascade that produces thrombin • Thrombin also converts fibrinogen to fibrin cementing platelets in place Thrombocytopenia • Spontaneous bleeding, prolonged bleeding time • Lowered platelet count – Uremia, too much aspirin and rare genetic disorders – Various marrow failure or injury • Aplastic anemia, leukemia – Sometimes immunologically mediated – Destruction of platelets by prosthetic heart valves • Bleeding into CNS a concern • Common hematologic manifestation of AIDS Coagulation Cascade • A series of conversions of inactive proenzymes to activated enzymes, – culminating in the formation of thrombin • Thrombin then coverts the soluble plasma protein fibrinogen to insoluble fibrous protein fibrin Two pathways of coagulation cascade • Intrinsic – Surface contact • Extrinsic – Tissue injury Coagulation cascade Hemophilia A (classic) is due to reduced amount or reduced activity of Factor VIII Hemophilia B (Christmas Disease) results from Deficiency of factor IX Heparin is a cofactor that Allows antithrombin III to Inactivate thrombin and Factor Xa Thrombomodulin binds To thrombin, making it An anticoagulant which Then activates antiCoagulant protein C. Protein C cleave factors Va and VIIIa Control of cascade to prevent clotting elsewhere • Antithrombins • Plasminogen– activated by heparin like plasmin system molecules on endothelial cells • Clinical administration of heparin minimizes thrombosis • Proteins C and S – Vitamin K dependent – Inactivate cofactors Va and VIIIa – Breaks down fibrin and inhibits its polymerization – Products of split fibrin are anticoagulants Conditions Causing Bleeding • Incomplete hemostasis is most common cause of bleeding. • Vitamin K deficiency – severe coagulation defect – Required for synthesis of prothrombin and factors VII, IX and X • Parenchymal diseases of the liver – Liver synthesizes several coagulation factors Hereditary deficiencies • Hemophilia A--factor VIII deficiency – Sex-linked recessive – 30% due to new mutations and don’t have family link – Hemarthroses common (spontaneous bleeding in joints) – Infuse patient with factor VIII from human blood or cryoprecipitate. – Need 100% levels preop, keep at 30% postop. • Hemophilia B--factor IX deficiency – Clinically indistinguishable from Hemophilia A – Sex-linked recessive – Need 50% preoperatively – Prolonged PTT, normal PT TX: Factor IX or FFP Von Willebrand’s Disease Von Willebrand’s Disease- Most common • • • • • congenital bleeding disorder. Types I, II, and III. PT normal PTT normal or elevated Prolonged bleeding time Type I most common (70%) with mild sx. Type III causes most bleeding • Type I and IIIreduced quantity of vWF • TX: Cryo, DDAVP • Type II- defect in vWF molecule, enough of it but doesn’t work well. TX: Cryo Platelet Disorders • Acquired-H2 blockers, heparin. • Bernard-Soulier, a Gp1b receptor deficiency (can’t bind to each other) • Uremia-Inhibits Gp1b, vWF. TX: dialysis, DDAVP, cryo, plts. • Ticlopidine- decreases ADP in platelets, prevents exposure of Gp1b/IIIa receptors. • Dipyramidoledecreases ADP induced plt aggregation. • Plavix-ADP receptor antagonist. • Pentoxifyllin- inhibits plt aggregation Platelet Disorders • Heparin induced thrombocytopenia (HIT) • Antiplatelet antibodies results in platelet destruction. • Also causes aggregation and thrombosis. • Low doses of heparin. • LMWH may have less risk. • DIC • Decreased plts, prolonged PT, PTT. • Low fibrinogen, high fibrin split products, high D-dimers • Often initiated by tissue factor. • Treat underlying cause Thrombi • Platelet aggregates • Fibrin • Trapped erythrocytes and leucocytes – Not part of this hemostatic process • This is a deep vein thrombosis – Postoperative patients confined to bed Thrombotic cycle and influences Endothelial injury is dominant influence • Heart and arterial circulation • Hypercholesteremia, cigarette smoke products • Radiation, bacterial endotoxins • Results in exposure of subendothelial collagen, adherence of platelets, release of tissue factor and local depletion of prostacyclin (antithrombin) Alterations in normal blood flow • Turbulence – injures endothelium – Atherosclerotic plaques • Stasis – Can create venous thrombi – Aneurysms cause local stasis – Deformed cells of Sickle cell anemia cause vascular occlusions Stasis and Turbulence • Disrupt laminar flow (cells in middle) • Bring platelets into contact with endothelium • Prevent dilution of activated clotting factors by fresh-flowing blood • Retard inflow of clotting factor inhibitors Hypercoagulability • Leiden Factor- 30% spontaneous venous thrombosis. • Most common congenital disorder. • Resistance to Protein C, defect on factor V • TX: heparin, warfarin. • Protein C, S deficiency-5% venous thromboses. TX: heparin, warfarin. Hypercoagulability • Antithrombin III deficiency- 2-3% thrombosis. Heparin doesn’t work. Can develop after previous heparin exposure. • TX: ATIII concentrate, FFP(highest conc) followed by heparin. • Polycythemia- defect in platelet function, usually thrombotic, but can bleed. Keep HCT<48, PLTS<400 before SX. TX: ASA. Hypercoagulability • Lupus Anticoagulant- antiphospholipid antibodies. Not all patients have SLE. A procoagulant (prolonged PTT but hypercoagulable). • Diagnose by seeing prolonged PTT, false positive RPR test for syphilis. • TX: heparin, warfarin. Hypercoagulability • Cardiopulmonary Bypass- factor XII activated, results in hypercoagulable state. TX: heparin, warfarin. • Warfarin induced skin necrosis- Occurs when coumadin started before heparin. Due to a decrease in protein S,C making patient transiently hypercoagulable. Patients with Protien C deficiency highly susceptible. Hypercoagulability • May be genetic or acquired • Genetic mutations in the factor V gene – Mutant factor V is resistant to anticoagulant effect of activated protein C • Smoking, obesity and age • Late pregnancy and postpartum – amniotic fluid infusion into circulation • Disseminated cancers – tumors release procoagulants • Advanced age, bed rest and immobilization Hematologic Drugs • Warfarin- prevents Vit K dependent decarboxylation of glutamic residues on Vit K dependent factors. • Dextran- inhibits platelets and coagulation factors. • Sequential compression devices-improve venous return but also induce fibrinolysis with compression (release of tPA). Hematologic Drugs • Heparin- activates antithrombin III. • Reversed with protamine (1-1.5 protamine per 100u of heparin). • Half-life 60-90 min. • Long term heparin use causes osteoporosis, alopecia. Does not cross BBB. • Protamine side-effects are hypotension, bradycardia, decreased heart function. Hematologic Drugs • Hirudin- leeches, thrombin inhibitor. • Ancrod- Malayan pit viper venom, stimulates tPA release. • Amicar-antifibrinolytic, procoagulant, inhibits plasmin. Used in DIC, persistent bleeding following CABG, thrombolytic overdoses. • Streptokinase, tPA-need to follow fibrinogen levels, levels<100 associated with severe bleeding. Fate of thrombus • Propagation – Accumulate more platelets and fibrin and obstruct a critical vessel • Embolization – Dislodge thrombi and transported to other sites in vasculature Fate of thrombus, continued • Dissolution/resolution – Removed by fibrinolytic activity • Organization – Induce inflammation and fibrosis and may reopen and allow blood flow Fates continued • Recanalization – Openings and even blood vessels created in thrombus