Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
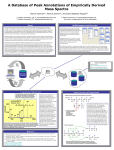
A Data Model for Annotating the Peaks of Mass Spectrum Data Dennis Harman[1] David Sigfredo Angulo[2] Kevin Drew[3] Alex Schilling[4] [1] DePaul University, CTI. [email protected] [3] DePaul University, [email protected] (to whom correspondence is to be addressed) [4] University of Illinois at Chicago. [email protected] DePaul University, CTI. [email protected] [2] Mass Spectrometry Data The advent of mass spectrometry (MS) for characterization of proteins has revolutionized proteomics. MS is technology used to study gas-phase ions by mass analysis, i.e., the separation of ions by their m/z (mass/charge) values. Although various mass spectrometers differ in their sensitivity and technology used in calculating m/z, all of them generate mass spectra as output. The raw data generated by a mass spectrometer must be refined before being informative. The process involves the interpretation of a mass spectrum. This requires a knowledge of the protonated peptide structure and the way that a peptide fragments [1]. The terminology conventionally used in describing MS ions encapsulates this information. Descriptive information using such terminology can be added to a mass spectrum as metadata. Summary MS protein sequencing techniques can provide information which allows researchers to determine the amino acid sequence of peptides. A data model is described that will allow for the annotation of mass peak data produced by a range of mass spectrometry experiments directed at peptide sequencing, characterization of post-translational modifications, and metabolite profiling. The model includes comprehensive sets of commonly used ion fragmentation terminology, while also allowing the user flexibility by providing userdefined text fields. The next phase of work will involve completing a physical data design of the model that integrates into the current IBG-MSP database schema and implementing memory-resident data structures to represent annotations. Entity-Relationship Model Components and Data Flows in the Annotation Process Process Workflow for a Mass Spectrometry-based Proteomics Experiment Analysis of a peptide mass spectrum provides information about the amino acid sequence of the peptide contained in the spectrum. Ions are displayed as spectral peaks, which can be annotated, e.g., a mass peak may be annotated as a “y3” ion. Data requirements for our annotations data model were determined by identifying possible ion types and focusing on use case scenarios to identify other forms of data that a researcher using MS may want to include as annotations to a mass spectrum. In addition to annotations useful for peptide sequencing experiments, the model also allows for annotating experimental data concerned with the characterization of posttranslational modifications of proteins, and the identification of metabolites (intermediate molecules resulting from chemical reactions occurring within biological systems). The annotations data model is mapped to an XML schema that will extend mzXML, an open, XML-based representation for MS data [2]. The XML model will support data exchange between the relational Illinois Bio-Grid Mass Spectrum database (IBG-MSD) [3] and other tools. One such tool is the IBG Desktop, an opensource tool which can be used to edit and display annotations [4]. *Ruedi, A., Matthias M., "Mass spectrometry-based proteomics", Nature vol. 422, 2003 March, pp. 198-207. Excerpt from XML Schema <?xml version="1.0" encoding="UTF-8"?> <xs:schema xmlns:xs="http://www.w3.org/2001/XMLSchema" elementFormDefault="qualified"> <xs:element name="peakAnnotations"> <xs:complexType> <xs:sequence> <xs:element minOccurs="0" maxOccurs="unbounded" ref="annotation"/> </xs:sequence> </xs:complexType> </xs:element> <xs:element name="annotation"> <xs:complexType> <xs:sequence> <xs:element minOccurs="0" ref="ionSeriesType"/> </xs:sequence> <xs:attribute name="id" use="required"/> <xs:attribute name="mz" use="required"/> <xs:attribute name="intensity" use="required"/> <xs:attribute name="comment"/> </xs:complexType> </xs:element> References [1] M. Kinter and N.E. Sherman, Protein Sequencing and Identification Using Tandem Mass Spectrometry, 2000; John Wiley & Sons, Inc, New York, NY. [2] P.G.A. Pedrioli1, et. al., “A common open representation of mass spectrometry data and its application to proteomics research”, Nature Biotechnology, vol. 22, 2004 Nov, pp. 1459-1466. [3] http://histone.cti.depaul.edu/IBG-MSP/login.do [4] http://gridweb.cti.depaul.edu/twiki/bin/view/IBG/IBGSoft#IBG_DeskTop Acknowledgements Work supported in part by the National Institute of Health (NIH) under Grant No. R01 HG3864.