Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
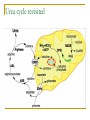
Ornithine Transcarbamylase Deficiency (OTCD) 9/1/09 Justin A. Crocker, MD What is it? X-linked disorder of the mitochondrial urea cycle Occurs 1 in every 80,000 people. associated with hyperammonemia affecting mainly male patients. Frequently, they die during the neonatal period, unless an early diagnosis and intensive management are carried out. Some patients with partial enzyme deficiencies might be asymptomatic until adulthood Heterozygous female patients exhibit a wide range of clinical manifestations. Urea cycle revisited Pathophys OTCD leads to the impaired condensation of carbamyl phosphate and ornithine to form citrulline. This impairment leads to reduced ammonia incorporation which, in turn, causes symptomatic hyperammonemia and excess of both substrates for the reaction. Under normal circumstances urea cycle occurs in hepatic mitochondria. However when excessive carbamyl phosphate it finds its way into the cytosol where it functions as substrate for the carbamoyl phosphate synthetase (CPS) reaction. This results in orotic acid, which is a normal intermediate in the very tightly regulated pyrimidine biosynthesis. In OTCD neither conversion of CPS to orotate nor hepatic leakage of ornithine can prevent the rapid development of hyperammonemia. Glutamine accumulation in the brain from hyperammonia is likely culprit of cerebral edema. Causes Mechanisms that can tip the nitrogen balance appear to break down into three categories: (1) nitrogen turnover and nitrogen load from catabolism or sudden protein processing (2) diminished access to processing in the liver (3) the genetic capacity of the urea cycle to handle the nitrogen load Nitrogen turnover and nitrogen load Rapid weight loss and poor nutritional intake Gastric bypass surgery Internal bleeding or damage Fracture Surgical damage Viral illness or other generalized stress Postpartum period Dramatic increase/decrease in habitual protein intake High protein diet strategies Change in food access or preparation Malabsorption conditions Medications affecting protein catabolism Intravenous or high-dose glucocorticoids Chemotherapy Diminished access to processing in the liver Vascular shunting of blood from liver Portacaval shunt Varicocele shunting from cirrhotic liver Decrease in available hepatocyte pool Chronic cirrhosis Acute chemical or viral damage to the liver Genetic predisposing conditions Genetic defects in enzyme/transporter function of the urea cycle components or decreased function polymorphisms Chemical or toxic affect on enzyme function 5-pentanoic acid (Jamaican vomiting sickness) Valproic acid Chemotherapeutic agents (cyclophosphamide) Comorbid metabolic conditions Organic acidemias Fatty acid oxidation defects Diagnosis Family history, dietary history, episodic nonspecific symptoms, response to withdrawal of protein Evaluate for possible precipitating events as outlined above: GI bleeding, trauma, viral illness, pregnancy test, offending medications, liver disease Plasma amino acids and urine organic acids Genotyping with skin fibroblasts Management Physically remove NH3 Usually done by dialysis or some form of hemofiltration. Begin at the highest available flow rate as soon as possible. Dialysis is the most effective means of ammonia removal, and the rate is dependent on the flow through the dialysis circuit In severe cases of hyperammonemia, provision for hemofiltration following the dialysis should be made until the patient is stabilized and the catabolic state is reversed. Some patients will reaccumulate ammonia after their initial round of dialysis and may require additional runs. Most patients will have a slight rise in ammonia after dialysis because removal by scavengers and the liver will not be as effective. This slight rise usually does not necessitate repeat dialysis. Reverse the catabolic state Fluids, dextrose, and fat emulsion (Intralipid) should be given. Insulin and glucose to artificially suppress the catabolic state is useful in profound catabolic states protein should be temporarily removed from diet Supplementation of arginine serves to replace arginine not produced by the urea cycle and prevents its deficiency from causing additional protein catabolism. Pharmacologic scavenging of excess nitrogen. Loading dose followed by a maintenance infusion of sodium benzoate/sodium phenylacetate Sodium benzoate combines with glycine to make hippurate, which is excreted by the kidneys Sodium phenylacetate combines with glutamine to make phenacetylglutamine, which is also excreted in the urine. The body replaces these amino acids using excess nitrogen. Additional issues Use of osmotic agents such as mannitol is not felt to be effective in treating the cerebral edema of hyperammonemia IV steroids and valproic acid should be avoided Antibiotics and a septic work-up are indicated to treat potential triggering events Rapid response to the hyperammonemia improves outcome. Symptomatology centers around cerebral edema and pressure on the brain stem. Long-term Avoid similar triggering events. In particular, intravenous steroids for asthma or valproic acid are contraindicated. Long-term diet modification with nutritional oversight is often necessary Avoid dehydration Special precautions must be taken to avoid catabolism during subsequent illnesses or surgeries, as well as during any event resulting in significant bleeding or tissue damage. It is important to provide genetic counseling to assess risk to other family members. References Bajay SK, Kurlemann G, Schuierer G, et al. CT and MRI in a girl with late-onset ornithine transcarbamylase deficiency: case report. Neuroradiology. 1996;38:796-799. Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology and therapy. Advanced Pediatrics. 1996;43:127170. Lien, J, Nyhan, WL, et. al. Fatal Initial Adult-Onset Presentation of Urea Cycle Defect, Archives of Neurology. 2007;64(12):17771779. Summar, ML, et. Al. Unmasked Adult-Onset Urea Cycle Disorders in the Critical Care Setting. Critical Care clinics. 2005;21. Uptodate