Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Ancestral sequence reconstruction wikipedia , lookup
Western blot wikipedia , lookup
Metalloprotein wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Interactome wikipedia , lookup
Protein purification wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Ioerger Lab – Bioinformatics Research
• Pattern recognition/machine learning
– issues of representation
– effect of feature extraction, weighting, and interaction on
performance of induction algorithm
• Applications in Structural Biology
–
–
–
–
–
–
molecular basis of biology: protein structures
predicting structures
tools for solving structures (X-ray crystallography, NMR)
stability, folding, packing, motions
drug design (small-molecule inhibitors)
large datasets exist – exploit them – find the patterns
TEXTAL - Automated Crystallographic Protein Structure
Determination Using Pattern Recognition
Principal Investigators:
Thomas Ioerger (Dept. Computer Science)
James Sacchettini (Dept. Biochem/Biophys)
Other contributors:
Tod D. Romo, Kreshna Gopal, Erik McKee,
Lalji Kanbi, Reetal Pai & Jacob Smith
Funding: National Institutes of Health
Texas A&M University
X-ray crystallography
• Most widely used method for
protein modeling
• Steps:
– Grow crystal
– Collect diffraction data
– Generate electron density map
(Fourier transform)
– Interpret map i.e. infer atomic
coordinates
– Refine structure
• Model-building
– Currently: crystallographers
– Challenges: noise, resolution
– Goal: automation
X-ray crystallography
• Most widely used method for
protein modeling
• Steps:
– Grow crystal
– Collect diffraction data
– Generate electron density map
(Fourier transform)
– Interpret map i.e. infer atomic
coordinates
– Refine structure
• Model-building
– Currently: crystallographers
– Challenges: noise, resolution
– Goal: automation
Overview of TEXTAL
• Automated model-building program
Electron density map
(or structure factors)
TEXTAL
Protein model
(may need refinement)
• Can we automate the kind of visual processing of
patterns that crystallographers use?
– Intelligent methods to interpret density, despite noise
– Exploit knowledge about typical protein structure
• Focus on medium-resolution maps
– optimized for 2.8A (actually, 2.6-3.2A is fine)
– typical for MAD data (useful for high-throughput)
– other programs exist for higher-res data (ARP/wARP)
Crystal
Collect data
Diffraction data
Electron density map
CAPRA: models backbone
LOOKUP: model side chains
SCALE MAP
TRACE MAP
Model of backbone
CALCULATE
FEATURES
PREDICT Cα’s
Model of backbone & side chains
BUILD CHAINS
PATCH & STITCH
CHAINS
POST-PROCESSING
SEQUENCE ALIGNMENT
REFINE CHAINS
REAL SPACE REFINEMENT
Corrected & refined model
F=<1.72,-0.39,1.04,1.55...>
F=<0.90,0.65,-1.40,0.87...>
F=<1.58,0.18,1.09,-0.25...>
F=<1.79,-0.43,0.88,1.52...>
Examples of Numeric Density Features
•Distance from center-of-sphere to centerof-mass
•Moments of inertia - relative dispersion
along orthogonal axes
•Geometric features like “Spoke angles”
•Local variance and other statistics
Features are designed to be rotation-invariant, i.e. same
values for region in any orientation/frame-of-reference.
TEXTAL uses 19 distinct numeric features to represent
the pattern of density in a region, each calculated over
4 different radii, for a total of 76 features.
The LOOKUP Process
Find optimal
rotation
Database
of known
maps
Region in map to
be interpreted
Two-step filter:
1) by features
2) by density
correlation
“2-norm”: weighted Euclidean
distance metric for retrieving matches:
dist ( R1 , R2 )
w (F (R ) F (R
i
i
i
1
i
2
)) 2
SLIDER: Feature-weighting algorithm
•
Euclidean distance metric used for retrieval:
dist ( R1 , R2 )
w (F (R ) F (R
i
i
1
i
2
)) 2
i
• relevant features – good, irrelevant features – bad
• Goal: find optimal weight vector w the generates highest
probability of hits (matches) in top K candidates from database
•
Concept of Slider:
• adjust features so the most matches are ranked higher than mismatches
Slider Algorithm(w,F,{Ri},matches,mismatches)
choose feature fF at random
for each <Ri,Rj,Rk>, Rjmatches(Ri),Rkmismatches(Ri)
compute cross-over point li where:
dist’(Ri,Rj)=dist’(Ri,Rk)
dist’(X,Y)= l(Xf-Yf)2+(1-l)dist\f(X,Y)
pick l that is best compromise among li
ranks most matches above mismatches
update weight vector: w’update(w,f,l), wf’=l
repeat until convergence
Quality of TEXTAL models
• Typically builds >80% of the protein
atoms
• Accuracy of coordinates: ~1Å error
(RMSD)
– Depends on resolution and quality of map
Closeup of b-strand (TEXTAL model in green)
Deployment
• September 2004: Linux and OSX distributions
– Can be downloaded from http://textal.tamu.edu
– 40 trial licenses granted so far
• June 2002: WebTex (http://textal.tamu.edu)
–
–
–
–
Till May 2005: TB Structural Genomics Consortium members only
Recently open to the public
users upload data; processed on server; can download results
120 users from 70 institutions in 20 countries
• July 2003: Model building component of PHENIX
– Python-based Hierarchical ENvironment for Integrated Xtallography
– Consortium members:
• Lawrence Berkeley National Lab
• University of Cambridge
• Los Alamos National Lab
• Texas A&M University
Intelligent Methods for Drug Design
• structure-based:
– given protein structure, predict ligands
that might bind active site
• other methods:
– QSAR, high-throughput/combi-chem,
manual design using 3D
• Virtual Screening
– docking algorithm + large library of
chemical structures
– sort compounds by interaction energy
– purchase top-ranked hits and assay in lab
– looking for mM inhibitors (leads that can
be refined)
– goal: enrichment to ~5% hit rate
Virtual Screening
• diversity
• ZINC database: ~2.6 million compounds
– purchasable; satisfy Lipinski’s rules
• docking algorithms:
– FlexX, DOCK, GOLD, AutoDock, ICM...
– search for position and conformation of ligand
• scoring function
– electrostatic + steric + desolvation
– entropy effects?
• major open issues:
– active site flexibility, charge state, waters, co-factors
– works best with co-crystal structures (already bound)
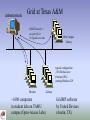
gridmaster.tamu.edu
Grid at Texas A&M
DOCK binaries +
receptor files +
20 ligands at a time
West Campus
Library
typical configuration:
2.8 GHz dual-core
Pentium CPUs
running Windows XP
Blocker
~1600 computers
in student labs on TAMU
campus (Open-Access Labs)
Zachary
GridMP software
by United Devices
(Austin, TX)
Data Mining of Results
•
•
•
•
•
promiscuous binders
clusters of related compounds
patterns of contacts within active site
hydrogen-bonding interactions
adjust weights of scoring function for unique
properties of each site
– open/closed, hydrophobic/charged...
• ideas for active site variations
• development of pharmacophore search patterns
Current Screens in Sacchettini Lab
• proteins related to tuberculosis (Mycobacterium)
– focus on unique pathways involved in dormancy/starvation
• glyoxylate shunt – slow-growth metabolic pathway
• cell-wall biosynthesis (unique mycolic acid layer in tb.)
• biosynthesis of amino acids/co-factors that humans get from diet
–
–
–
–
–
–
–
–
isocitrate lyase
malate synthase
PcaA: mycolic acid cyclopropane synthase
ACPS: acyl-carrier protein synthase
InhA: enoyl-acyl reductase (target of isoniazid)
KasB: fatty-acid synthase
BioA: biotin (co-factor) synthase
PGDH: phospho-glycerol dehydrogenase (serine biosynthesis)
• Related proteins in malaria, SARS, shigella
Conclusions
• Many opportunities for research in Structural Bioinformatics
– large datasets
– significant problems
• Provides challenges for machine learning
– drives development of novel methods, especially for dealing with noise,
sampling biases, extraction of features...
• Requires inherently interdisciplinary approach
– training in biochemistry; knowledge of molecular interactions
– understanding chemical intuition; use of visualization tools
– insights about strengths and limitations of existing methods
• Requires collaboration to construct appropriate representations to
enable learning algorithms to find patterns
– translate expectations about what is relevant, dependencies, smoothing,
sources of noise...