Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Discovery and development of cyclooxygenase 2 inhibitors wikipedia , lookup
Discovery and development of non-nucleoside reverse-transcriptase inhibitors wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Compounding wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Psychopharmacology wikipedia , lookup
Drug design wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacognosy wikipedia , lookup
Drug discovery wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Plateau principle wikipedia , lookup
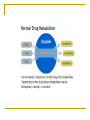
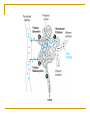
Drug elimination (metabolism, excretion) Anton Kohút DRUG METABOLISM major site of drug metabolism – liver, exceptions: - suxamethonium and procaine - plasma cholinesterase - tyramine - intestinal wall Phase 1 - lipophilic molecules are converted into more polar molecules Reactions are catalyzed by the cytochrome P-450 (CYP) by: oxidation reduction hydrolysis - products are often less eactive than the parent compounds - after metabolism may be excreted by kidneys - each of the enzyme has a low specificity Phase 2 consists of conjugation reactions Most often involved groups in conjugation are: glucuronyl sulphate methyl acetyl glycyl glutamyl The two phases of drug metabolism Metabolism of imipramine Involvement of some isoform of CYP in the metabolism FACTORS INFLUENCING DRUG METABOLISM - systemic pathological processes - liver diseases, heart failure - age - sex - body temperature - genetic factors - polymorhism - drug interactions enzyme inhibition, enzyme induction Drug interactions - enzyme induction Drug induction increase of enzyme activity can decrease drug potency, - result is an increased synthesis of microsomal enzymes after repeated use of drugs - generally, metabolism of inducers itself is increased as well as various other compounds, - increase of metabolism may increase toxicity of paracetamol – toxic metabolites Drugs that cause induction barbiturates, carbamazepine, ethanol (chronic use), glutethimide, griseofulvin, meprobamate, phenytoin, rifampicin, sulphinpyrazone, Drug interactions - enzyme inhibition Drug inhibition - enzyme inhibition can slow down the metabolism - action of coadministrated drug may be increased and prolonged Drugs that cause inhibition - cimetidine, - erythromycin, - quinolone - sodium valproat, - allopurinol Genetic polymorphism Genetic polymorphism Genetic polymorphism 1. Plymorphism in acetylation (rapid or slow acetylators – isoniasid (INH), hydralazine and procainamide, sulphasalazine 2. Poor oxidisers – debrisoquine, metoprolol, timolol, haloperidol 3. Glucose-6-phosphate dehydrogenase deficiency G-6-PD – risk of haemolysis – aspirin, probenecid, quinine, chloroquine, nitrofurantoin, some sulphonamides 4. Pseudocholinesterase deficiency – malignant hyperthermia – suxametonium, CYP 2D6 Drug excretion Drug excretion - Drugs are exreted : either unchanged or as metabolites. - Lipid-soluble drugs are not readily eliminated until they are metabolized to more polar compounds Renal excretion Extrarenal excretion Renal excretion 1. Glomerular filtration - passive Depends on: fractional plasma protein binding glomerular filtration rate (size of molecules) (MW 68 000) is completely held back by this way is removed about 20% of drugs from the blood 2. Passive tubular reabsorption the nonionized forms of weak acids and bases undergo passive reabsorption. The concentration gradient for back-diffusion is created by the reabsorption of water with Na+ and other inorganic ions. When the tubular urine is made more acidic, the excretion of weak acids is reduced (alkalinization of the urine have the opposite effects on the excretion of weak bases). Treatment of drug poisoning, the excretion of some drugs can be hastened by appropriate alkalinization or acidification of the urine. Renal excretion (cont.) 3. Active tubular secretion Many organic acids (such as penicillin) and metabolites (such as glucuronides) are transported by the system that secretes naturally occurring substances (such as uric acid). Organic bases, such as tetraethylammonium, are transported by a separate system that secretes choline, histamine, and other endogenous bases. The carrier systems are relatively nonselective, and organic ions of similar charge compete for transport. Both transport systems also can be bidirectional, and at least some drugs are both secreted and actively reabsorbed. (an endogenous organic acid is uric acid). Excretion by other routes Biliary and fecal excretion Saliva, sweat, tears - the concentration of some drugs in saliva parallels that in plasma. Breast milk - excretion by breast milk is dependent mainly upon diffusion, Milk is more acidic than plasma, basic compounds may be slightly concentrated in this fluid. Excretion by Other Routes 1. Saliva Excretion of drugs into sweat, saliva, and tears is quantitatively unimportant. Excretion by saliva is dependent mainly upon diffusion Drugs excreted in the saliva enter the mouth, where they are usually swallowed. The concentration of some drugs in saliva parallels that in plasma. Saliva may therefore be a useful biological fluid in which to determine drug concentrations when it is difficult or inconvenient to obtain blood. 2. Breast milk Excretion by breast milk is dependent mainly upon diffusion Milk is more acidic than plasma, basic compounds may be slightly concentrated in this fluid, Nonelectrolytes, such as ethanol and urea, readily enter breast milk and reach the same concentration as in plasma, independent of the pH of the milk. Excretion of drugs in breast milk are potential sources of unwanted pharmacological effects in the nursing infant. 3. Feces Substances excreted in the feces are mainly unabsorbed orally ingested drugs or metabolites excreted in the bile and not reabsorbed from the intestinal tract. 4. Biliary excretion Elimination Rate of elimination Elimination of most drugs from the body after therapeutically relevant doses follows first-order kinetics. To illustrate first order kinetics we might consider what would happen if we were to give a drug by i.v. bolus injection, collect blood samples at various times and measure the plasma concentrations of the drug. We might see a decrease in concentration as the drug is eliminated. First-order kinetics Elimination half-life (t1/2) Definition: Elimination half-life is the time it takes the drug concentration in the blood to decline to one half of its initial value. It is a secondary parameter : The elimination half-life is dependent on the ratio of VD and CL. Unit : time (min, h, day) First order kinetics half - life Most drugs exhibit first order kinetics – disappeaance of drug from plasma follows exponential patterns. The rate of elimination is directly proportional to drug concentration. Plasma half-life is directly proportional to the volume of distribution and inversely proportional to the overall rate of clearance With repeated dosage or sustained delivery of a drug the plasma concentration approaches a steady state within 4-5 half-lifes. Rate of elimination Elimination which follows first-order kinetics: dC/dt = k . C el Dose=100 mg, V =10 L, Conc. (mg/L) 10 d C0=Dose/Vd = 10 mg/L 7.5 5 5 2.5 2.5 1.25 0.625 0 0 2 4 6 time (h) 8 10 kel …. rate constant of elimination rate of change is proportional to concentration and is therefore decreasing with time as the conc. decreases Rate of elimination monoexponential decay: C(t) = C0 . e- kel . t Conc. (mg/L) half-life : t1/2 Dose=100 mg, Vd=10 L, C0=Dose/Vd = 10 mg/L 10 7.5 C= C0 / 2 5 5 t= t1/2 = ln2 / kel = 0.693/ kel 2.5 2.5 1.25 0.625 0 after 4 half-lives: 0 2 4 6 time (h) 8 10 6% remaining, 94% eliminated Saturation kinetics In a few cases as ethanol, phynytoin and salicylate disapppearance of drug from plasma does not follow exponential patterns. Pattern of desappearance of drug is linear – drug is removed at a constant rate that is indipendent of plasma concentration This is often called zero order kinetics Use of t1/2: 1/ t1/2 can be used to predict how long it will take for the drug to be eliminated from plasma. 1. Conc. (mg/L) 10 2. 50 7.5 5 4. 3. 5. 87.5 94 97 75 percent eliminated % t1/2 = 2 hours 5 2.5 2.5 1.25 0.625 0 0 2 4 6 time (h) 8 10