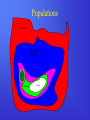
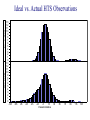
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Problems and Opportunities for Machine Learning in Drug Discovery (Can you find lessons for Systems Biology?) George S. Cowan, Ph.D. Computer Aided Drug Discovery Pfizer Global Research and Development, Ann Arbor Labs CSSB, Rovereto, Italy 19 April 2004 Working as a Computer Scientist in a Life Sciences field requires an array of supporting scientists Thanks to: Cheminformatics Mentors and Colleagues: John Blankley Alain Calvet David Moreland Academic: Peter Willett Robert Pearlman Project Colleagues: David Wild Kjell Johnson Risk Takers: Eric Gifford Mark Snow Christine Humblet Mike Rafferty Drug Discovery and Development Discern unmet medical need Discover mechanism of action of disease Identify target protein Screen known compounds against target Synthesize promising leads Find 1-2 potential drugs Toxicity, ADME Clinical Trials Lock and Key Model Virtual HTS Screening Virtual Screening Definition • estimate some biological behavior of new compounds • identify characteristics of compounds related to that biological behavior • only use some computer representation of the compounds HTS Virtual Screening is Not QSAR/QSPR • Based on large amounts of easy to measure observations • Uses early stage data from multiple chemical series (no X-ray Crystallography) • Observations are not refined (Percent Inhibition at a single concentration) • Looking for research direction, not best activity Promise of Data Mining Data Mining • • • • Works with large sets of data Efficient Processing Finds non-intuitive information Methods do not depend on the Domain (Marketing, Fraud detection, Chemistry, …) Alternative Data Mining Approaches • • • • • Regression - Linear or Non-Linear - PLS Principal Components Association Rules Clustering Approach - Unsupervised - Concept Formation Classification Approach - Supervised Overview (1) Virtual Screening Challenges to Machine Learning • No single computer representation captures all the important information about a molecule • The candidate features for representing molecules are highly correlated • Features are entangled – Multiple binding modes use different combinations of features – Multiple chemical series / scaffolds use the same binding mode – Evidence that some ligands take on multiple conformations when binding to a target – Any 4 out of 5 important features may be sufficient More Challenges to Machine Learning • • • • • • Overview (2) Training data and validation data are not representative Measurements of activity are inherently noisy Activity is a rare event; target populations are unbalanced Classification requires choosing cutoffs for activity There is no good measure for a successful prediction Many data mining methods characterize activity in ways that are meaningless to a chemist • Data mining results must be reversible to assist a chemist in inventing new molecules that will be active (inverse QSAR) Deep Challenges to Machine Learning Overview (3) • No free lunch theorem • Science is different from marketing No Single Computer Representation captures all the important information How do we characterize the electronic “face” that the molecule presents to the protein? – Grid of surface or surrounding points with field calculations – Conformational flexibility – 3-D relationships of pharmacophores • Complementary volumes and surfaces • Complementary charges • Complementary hydrogen bonding atoms • Similar Hydrophbicity/Hydrophilicity – Connectivity: Bonding between Atoms (2-D) • pharmacophore info is implicitly present to some extent • not biased toward any particular conformation – Presence of molecular fragments (fingerprints) – Other: Linear (SLN, SMILES)? Free-tree? Pharmacophores Representation of Chemical Structures (2D) Aspirin BCI Chemical Descriptors • Descriptors are binary and represent - augmented atoms - atom pairs - atom sequences - ring compositions Chiral F O O N NH OH Ca 2+ OH OH We don’t have the right descriptors, but we have thousands that are easy to compute • Thousands of molecular fragments • Hundreds of calculated quasi-physical properties • Hundreds of structural connectivity indicators Much of this information is redundant Feature Interaction and Multiple Configurations for Activity Require Disjunctive Models • Multiple binding modes where different combinations of features contribute to the activity (including non-competitive ligands) • Multiple chemical series / scaffolds use the same binding mode • Any 3 out of 4 important features may be sufficient • Evidence that some targets require multiple conformations from a ligand in order to bind Non-competitive Binding Non-competitive Binding Unbalanced target populations (activity is a rare event) • About 1% of drug-like molecules have interesting activity • Most of our experience in classification methods is with roughly balanced classes • Predictive methods are most accurate where they have the most data (interpolation), but where we need the most accuracy is with the extremely active compounds (extrapolation) Warning: Your data may look balanced • True population of interest: – new and different compounds • Unrepresentative HTS training data: – What chemists made in the past • Unrepresentative follow-up compounds for validation: – What chemists intuition led them to submit to testing Populations All Drugs Possible with Current Technology Next next Library Tested Cipsline, Anti-infectives 2 Score HIV 1 0 -1 -2 -3 0 1000 2000 3000 4000 5000 6000 Our models are accurate on the compounds made by our labs Validation Statistics Depend on Prevalence of the Actives Count Column % Row % Accuracy = 0.792 Predicted Class Act Predictive Value = 0.936 Not_Act Actual Class Recall = 0.703 617 Act Not_Act 261 93.63 32.75 70.27 29.73 42 536 6.37 67.25 7.27 92.73 659 797 878 Sensitivity = 0.703 Specificity = 0.927 578 Kappa 0.592 1456 Std Err 0.0200 1 - Exp Kappa = ¾¾¾¾¾ Obs - Exp Redm an, C. E. “Screening Compounds for Clinically Act ive Drugs”, in Statis tics for the Pharm aceut ical Industry, 36, 19-42, 198 1 1% Prevalence Validation Statistics Actual Class Count Column % Row % Act Not_Act Accuracy = 0.925 Predicted Class Not_Act Predictive Value = 0.089 Recall = 0.703 703 297 Sensitivity = 0.703 8.90 0.32 70.27 29.73 7270 92,730 91.10 99.68 7.27 92.73 7900 92,100 Act 1000 Specificity = 0.927 Kappa = 0.147 99,000 100,000 NOTICE THAT Sensitivity and Specificity are equal to the previous slide but predictive value is much less Choosing cutoffs for activity and cutoffs for compounds to pursue • Overlapping ranges of Inactive and Active • Cost of missing an active vs. cost of pursuing an inactive Ideal vs. Actual HTS Observations 13 12 11 T 10 h 9 e o 8 r 7 e t 6 i c 5 a 4 l 3 2 1 0 13 12 11 10 O 9 b 8 s e 7 r v 6 e 5 d 4 3 2 1 0 -125 -105 -85 -65 -45 -25 -5 15 Percent Inhibition 35 55 75 95 115 135 ROC Curves 1 0.9 0.8 0.7 sensitivity 0.6 Cost 1/Ratio 0.5 IsoCost1 IsoCost2 0.4 IsoCost3 0.3 0.2 0.1 0 0 0.1 0.2 0.3 0.4 0.5 False Positive Rate 0.6 0.7 0.8 0.9 1 Virtual Screening # of active retrieved vs # of compounds tested 140 140 120 120 100 100 80 80 60 60 40 40 20 20 0 0 0 1000 2000 3000 4000 5000 1 10 100 # tested # tested Upper Ref # of active retrieved Random 1000 10000 We use the log-linear graph to compare methods at different follow-up levels See how 3 different methods perform at selecting 5, 50, or 500 compounds to test 130 110 100 80 RP 60 SOM 40 LVQ 20 Reference 0 2 20 200 2000 # of compounds screened 20000 Random Noise in measurement of activity • Suppose 1% active and 1% error, then our predicted actives are 50% false positives • This is out of the range of data-mining methods (but see “Identifying Mislabeled Training Data”, Brodley & Friedl, JAIR, 1999) • Luckily, the error in measuring inactives is dampened • Methods can take advantage of the accuracy in inactive information in order to characterize actives • On the other hand, inactives have nothing in common, except that they are the other 99% Mysterious Accuracy OR Neural Networks are great, but what are they telling me? We have a decision to make about data mining goals: • Do we try to: Outperform the chemist or engage the chemist We need to assist a chemist in inventing new molecules that will be active (inverse QSAR) We need to characterize activity in ways that are meaningful to a chemist No Free Lunch Theorem • Proteins recognize molecules • Proteins compute a recognition function over the set of molecules • Proteins have a very general architecture • Proteins can recognize very complex or very simple characteristics of molecules • Proteins can compute any recognition function(?) • No single data-mining/machine-learning method can outperform all others on arbitrary functions • Therefore every new target protein requires its own modeling method • “Cheap Brunch Hypothesis”: Maybe proteins have a bias Science, Not Marketing • We are looking for hypotheses that are worth the effort of experimental validation (not e-marketing opportunities) • Data-mining rules and models need to be in the form of a hypothesis comparable to the chemist’s hypotheses • Chemists need tools that help them design experiments to validate or invalidate these competing hypotheses • HTS is an experiment in need of a design Conclusion • Machine-learning tools provide an opportunity for processing the new quantities of data that a chemist is seeing • The naïve data-mining expert has a lot to learn about chemical information • The naïve chemist has a lot to learn about data-mining for information If there are so many problems why are we having so much fun? Maybe we’ve stumbled into the cheap brunch