Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
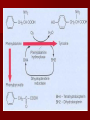
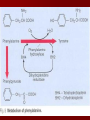
HEREDITARY METABOLIC DISEASES Particular risk factors are: •Advanced maternal age (e.g. Down's syndrome) • Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) • Previous child with genetic disorder (e.g. Tay-Sachs disease PHENYLKETONURIA Excess phenylalanine is normally converted to tyrosine, another amino acid, and eliminated from the body. Without the enzyme that converts it to tyrosine, phenylalanine builds up in the blood and is toxic to the brain, causing mental retardation. SYMPTOMS mental retardation over the first few years of life, which eventually becomes severe. Other symptoms include seizures, nausea and vomiting, an eczema-like rash, lighter skin and hair than their family members, aggressive or self-injurious behavior, hyperactivity, and sometimes psychiatric symptoms. Untreated children often give off a "mousy" body and urine odor as a result of a by-product of phenylalanine (phenylacetic acid) in their urine and sweat. MAPLE SYRUP URINE DISEASE By-products of lecine, isoleucine and valine build up, causing neurologic changes, including seizures and mental retardation. These by-products also cause body fluids, such as urine and sweat, to smell like maple syrup infants develop neurologic abnormalities, including seizures and coma, during the first week of life and can die within days to weeks In the milder forms, children initially appear normal but develop vomiting, staggering, confusion, coma, and the odor of maple syrup particularly during physical stress, such as infection or surgery Infants with severe disease are treated with dialysis. Some children with mild disease benefit from injections of the vitamin B1 (thiamin). After the disease has been brought under control, children must always consume a special artificial diet that is low in the particular amino acids that are affected by the missing enzyme. HOMOCYSTINURIA Children with homocystinuria are unable to metabolize the amino acid homocysteine, which, along with certain toxic by-products The first symptoms, including dislocation of the lens of the eye, causing severely decreased vision, usually begin after 3 years of age. Most children have skeletal abnormalities, including osteoporosis; the child is usually tall and thin with a curved spine, elongated limbs, and long, spiderlike fingers. In a few states, children are screened for homocystinuria at birth with a blood test. The diagnosis is confirmed by a test measuring enzyme function in liver or skin cells. TYROSINEMIA There are two main types of tyrosinemia: I and II. Type I tyrosinemia is most common in children of French-Canadian or Scandinavian descent. Children with this disorder typically become ill sometime within the first year of life with dysfunction of the liver, kidneys, and nerves, resulting in irritability, rickets, or even liver failure and death. Type II tyrosinemia is less common. Affected children sometimes have mental retardation and frequently develop sores on the skin and eyes. Unlike type I tyrosinemia, restriction of tyrosine in the diet can prevent problems from developing. GLYCOGEN STORAGE DISEASES There are many different glycogen storage diseases (also called glycogenoses), each identified by a roman numeral. These diseases are caused by a hereditary lack of one of the enzymes that is essential to the process of forming glucose into glycogen and breaking down glycogen into glucose. About 1 in 20,000 infants has some form of glycogen storage disease. GALACTOSEMIA Galactosemia (a high blood level of galactose) is caused by lack of one of the enzymes necessary for metabolizing galactose, a sugar present in lactose (milk sugar). A metabolite builds up that is toxic to the liver and kidneys and also damages the lens of the eye, causing cataracts. A newborn with galactosemia seems normal at first but within a few days or weeks loses his appetite, vomits, becomes jaundiced, has diarrhea, and stops growing normally. White blood cell function is affected, and serious infections can develop. If treatment is delayed, affected children remain short and become mentally retarded or may die. Types and Characteristics of Glycogen Storage Diseases Name Affected Organs Symptoms Type O Liver, muscle Enlarged liver with accumulation of fat inside the liver cells (fatty liver); episodes of low blood sugar levels (hypoglycemia) when fasting von Gierke's disease (Type IA) Liver, kidney Enlarged liver and kidney; slowed growth; very low blood sugar levels; abnormally high levels of acid, fats, and uric acid in blood Type IB Liver, white blood cells Same as in von Gierke's disease but may be less severe; low white blood cell count; recurring mouth and intestinal infections or Crohn's disease Pompe's disease (Type II) All organs Enlarged liver and heart, muscle weakness Forbes' disease (Type III) Liver, muscle, heart, white blood cells Enlarged liver or cirrhosis; low blood sugar levels; muscle damage and heart damage in some people Andersen's disease (Type IV) Liver, muscle, most tissues Cirrhosis in juvenile type; muscle damage and heart failure in adult (late-onset) type McArdle's disease (Type V) Muscle Muscle cramps or weakness during physical activity Hers' disease (Type VI) Liver Enlarged liver; episodes of low blood sugar when fasting; often no symptoms Tarui's disease (Type VII) Skeletal muscle, red blood cells Muscle cramps during physical activity; red blood cell destruction (hemolysis) Galactosemia is treated by completely eliminating milk and milk products—the source of galactose—from an affected child's diet. Galactose is also present in some fruits, vegetables, and sea products, such as seaweed. Doctors are not sure whether the small amounts in these foods cause problems in the long term. People who have the disorder must restrict galactose intake throughout life.