Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
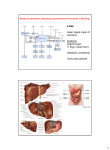
6.2.3 Liver As it is one of the portals of entry to the tissues of the body, the liver is exposed to many potentially toxic substances via the gastrointestinal tract from the diet, food additives and contaminants, and drugs and is frequently a target in experimental animals. In humans, liver damage is less common, and only around 9% of adverse drug reactions affect the liver. By virtue of its position, structure, function, and biochemistry, the liver is especially vulnerable to damage from toxic compounds. Substances taken into the body from the gastrointestinal tract are absorbed into the hepaticportal blood system and pass via the portal vein to the liver. Thus, after the gastrointestinal mucosa and blood, the liver is the next tissue to be exposed to a compound, and as it is prior to dilution in the systemic circulation, this exposure will often beat a higher concentration than that of other tissues. The liver, the largest gland in the body, represents around 2% to 3% of the body weight in humans and other mammals such as the rat. It is served by two blood supplies, the portal vein, which accounts for 75% of the hepatic blood supply, and the hepatic artery. The portal vein drains the gastrointestinal tract, spleen, and pancreas and therefore supplies nutrients, and the hepatic artery supplies oxygenated blood (Fig. 6.2). The liver receives around 25% of the cardiac output, which flows through the organ at around 1 to 1.3 mL min_1 g_1 and drains via the hepatic vein into the inferior vena cava. In between the blood entering the liver via hepatic artery and portal vein and leaving via the hepatic vein, the blood flows through sinusoids (Fig. 6.3). Sinusoids are specialized capillaries with discontinuous basement membranes, which are lined with Kupffer cells and endothelial cells. There are large fenestrations in the sinusoids, which allow large molecules to pass-through into the interstitial space and into close contact with the hepatocytes (Fig. 6.4). The liver is mainly composed of hepatocytes arranged as plates approximately twocells thick, each plate bounded by a sinusoid (Fig. 6.4). The membranes of adjacent hepatocytes form the bile canaliculi into which bile is secreted. The bile canaliculi form a network, which feed into ductules, which become bile ducts (Fig. 6.3). The structural and functional unit of the liver is the lobule, which is usually described in terms of the hepatic acinus (Fig. 6.5), based on the microcirculation in the lobule. When the lobule is considered in structural terms, it may be described as either a classical or a portal lobule (see “Glossary”). The acinus comprises a unit bounded by two portal tracts and terminal hepatic or central venules, where a portal tract is composed of a portal venule, bile ductile, and hepatic arteriole (Fig. 6.5). Blood flows from the portal tract toward the central venules, whereas bile flows in the opposite direction. There are three circulatory zones in the acinus, with zone 1 receiving blood from the afferent venules and arterioles first, followed by zone 2, and finally zone 3. Thus, there will be metabolic differences between the zones because of the blood flow. Zone 1 will receive blood, which is still rich in oxygen and nutrients, such as fats and other constituents. The hepatocytes in zone 3, however, will receive blood, which has lost much of the nutrients and oxygen. Zone 1 approximates to the periportal region of the classical lobule and zone 3 to the centrilobular region. Zone 3, particularly where several acini meet, is particularly sensitive to damage from toxic compounds. The acinus is also a secretory unit, the bile it produces flowing into the terminal bile ductules in the portal tract. The close proximity of the blood in the sinusoids with the hepatocytes allows efficient exchange of compounds, both endogenous and exogenous, and consequently foreign compounds are taken up very readily into hepatocytes. For example, the drug propranolol is extensively extracted in the “first pass” through the liver. The liver is a target organ for toxic substances for four main reasons: 1. The large and diverse metabolic capabilities of the liver enable it to metabolize many foreign compounds, but as metabolism does not always result in detoxication, this may make it a target (see sects. 7.2.1 and 7.2.4 chap. 7). 2. The liver also has an extensive role in intermediary metabolism and synthesis, and consequently, interference with endogenous metabolic pathways may lead to toxic effects, as discussed in chapter 7 (see sects. 7.8.2 and 7.8.3). 3. The secretion of bile by the liver may also be a factor. This may be due to the biliary excretion of foreign compounds leading to high concentrations, especially if saturated, as occurs with the hepatotoxic drug furosemide. Alternatively, enterohepatic circulation can give rise to prolonged high concentrations in the liver. Interference with bile production and flow as a result of precipitation of a compound in the canalicular lumen or interference with bile flow may lead to damage to the biliary system and surrounding hepatocytes. 4. The blood supply ensures that the liver is exposed to relatively high concentrations of toxic substances absorbed from the gastrointestinal tract. The hepatocytes, or parenchymal cells, represent about 80% of the liver by volume and are the major source of metabolic activity. However, this metabolic activity varies depending on the location of the hepatocyte. Thus, zone 1 hepatocytes are more aerobic and therefore are particularly equipped for pathways such as the boxidation of fats, and they also have more GSH and GSH peroxidase. These hepatocytes also contain alcohol dehydrogenase and are able to metabolize allyl alcohol to the toxic metabolite acrolein, which causes necrosis in zone 1. Conversely, zone 3 hepatocytes have a higher level of cytochromes P-450 and NADPH cytochrome P450 reductase, and lipid synthesis is higher in this area. This may explain why zone 3 is most often damaged, and lipid accumulation is a common response (see “Carbon Tetrachloride,” for instance, chap. 7). The Kupffer cells are known to contain significant peroxidase activity and also acetyltransferase. The differential distribution of is enzymes may also be a factor in the localization of damage. There are various types of toxic response, which the liver sustains and which reflect its structure and function. Viewed simply, liver injury is usually due either to the metabolic capabilities of the hepatocyte or involves the secretion of bile. The various types of liver damage, which may be caused by toxic compounds, are discussed in the following sections. Fatty Liver (Steatosis) This is the accumulation of triglycerides in hepatocytes, and there are a number of mechanisms underlying this response as is discussed below (see the sect. “Mechanisms of Toxicity”). The liver has an important role in lipid metabolism, and triglyceride synthesis occurs particularly in zone 3. Consequently, fatty liver is a common response to toxicity, often the result of interference with protein synthesis, and may be the only response as after exposure tohydrazine, ethionine, and tetracycline, or it may occur in combination with necrosis as with carbon tetrachloride. It is normally a reversible response, which does not usually lead to cell death, although it can be very serious as is the case with tetracycline-induced fatty liver in humans. Repeated exposure to compounds, which cause fatty liver, such as alcohol, may lead to cirrhosis. The specific accumulation of phospholipids (phospholipidosis) can occur but it also occurs in other organs and tissues and will be discussed later in this chapter. Cytotoxic Damage Many toxic compounds cause direct damage to the hepatocytes, which leads to cell death and necrosis. This is a general toxic response, not specific for the liver, and there are undoubtedly many mechanisms, which underlie cytotoxicity, but most are still poorly understood. The mechanisms underlying cytotoxicity in general are discussed below, and several examples of hepatotoxins are discussed in more detail in chapter 7. The zone of the liver damaged may depend on the mechanism, but may also be the result of the microcirculation. Damage may be zonal, diffuse, or massive. For example, cocaine and allyl alcohol cause zone 1 (periportal) necrosis. With allyl alcohol, this is partly as a result of the presence of alcohol dehydrogenase and partly because this is the first area exposed to the compound in the blood. Conversely, carbon tetrachloride, bromobenzene, and paracetamol cause zone 3 (centrilobular) necrosis as a result of metabolic activation occurring primarily in that region (see chap. 7). Midzonal, zone 2 necrosis is less common, but has been described for the natural product ngaione and beryllium. Galactosamine causes diffuse hepatic necrosis (seechap. 7), presumably because it interferes with a metabolic pathway, which occurs in all regions of the liver lobule. The explosive trinitrotoluene (TNT) can cause massive liver necrosis. Ischemia may also be a component of cytotoxic damage, and consequently interference with liver blood flow by toxic compounds such as phalloidin, which causes swelling of the endothelial cells lining the sinusoids, may cause or contribute toward cytotoxicity. Other compounds cause liver necrosis because of biliary excretion. Thus, the drug furosemide causes a dosedependent centrilobular necrosis in mice. The liver is a target as a result of its capacity for metabolic activation and because furosemide is excreted into the bile by an active process, which is saturated after high doses. The liver concentration of furosemide therefore rises disproportionately (chap. 3, Fig. 34), and metabolic activation allows the production of a toxic metabolite (Fig. 6.6). The drug proxicromil (chap. 5, Fig. 11) caused hepatic damage in dogs as a result of saturation of biliary excretion and a consequent increase in hepatic exposure. Cholestatic Damage There are various types of interference with the biliary system, and this can lead to bile stasis or damage to the bile ducts, ductules, or canaliculi. In some cases, such as with chlorpromazine, damage to the hepatocytes may ensue. Thus, some foreign compounds, such as the antibiotic rifampicin, interfere with bilirubin transport and conjugation giving rise to hyperbilirubinemia. Other compounds, icterogenin, for example, cause bile stasis and bilirubin deposits in the canaliculi. This canalicular damage may also be accompanied by damage to hepatocytes, such as caused by chlorpromazine. This drug is a surface-active agent, which can reach high concentrations in the bile and so directly damage the lining cells. It can also cause precipitation of insoluble substances in the lumen of the canaliculi. Accumulation of bile and its constituent bile salts may indeed be the cause of damage, and some are surface-active agents. Consequently, if high concentrations are reached, the cells of the biliary system and hepatocytes exposed can be damaged. The secondary bile acid lithocholate will cause direct damage to the canalicular membrane, for example. Some compounds damage the bile ducts and ductules directly such as anaphthylisothiocyanate. The result of the destruction of bile duct-lining cells will be cholestasis as debris from the necrotic cells will block the ductules. Cirrhosis This is a chronic lesion resulting from repeated injury and subsequent repair. It may result from either hepatocyte damage or cholestatic damage, each giving rise to a different kind of cirrhosis. Thus, carbon tetrachloride will cause liver cirrhosis after repeated exposure, but also compounds, which do not cause acute necrosis, such as ethionine and alcohol may cause cirrhosis after chronic exposure. Vascular Lesions Occasionally toxic compounds can directly damage the hepatic sinusoids and capillaries. One such toxic compound is monocrotaline, a naturally occurring pyrrolozidine alkaloid, found in certain plants (Heliotropium, Senecio, and Crotolaria species). Monocrotaline (Fig. 7.7) is metabolized to a reactive metabolite, which is directly cytotoxic to the sinusoidal and endothelial cells, causing damage and occlusion of the lumen. The blood flow in the liver is therefore reduced and ischemic damage to the hepatocytes ensues. Centrilobular necrosis results, and the venous return to the liver is blocked. Hence, this is known as veno- occlusive disease and results in extensive alteration in hepatic vasculature and function. Chronic exposure causes cirrhosis. Liver Tumors Both benign and malignant liver tumors may arise from exposure to hepatotoxins and can be derived from various cell types. Thus, adenomas have been associated with the use of contraceptive steroids and exposure to aflatoxin B1, and dimethylnitrosamine can produce hepatocellular carcinomas, whereas vinyl chloride causes hemangiosarcomas derived from the vasculature (see chap. 7). Proliferation of Peroxisomes A response to exposure to certain , which occurs predominantly in the liver is the phenomenon of peroxisomal proliferation. Peroxisomes (microbodies) are organelles found in many cell types, but especially hepatocytes. Repeated exposure of rodents to certain types of foreign compound leads to an increase in the number of these organelles and an increase in the activities of various enzymes. As well as exposure in vivo, exposure of isolated hepatocytes in vitro will also lead to proliferation of peroxisomes, indicating that it is a cellular response. The function of the organelle is mainly the oxidative metabolism of lipids and certain other oxidative metabolic pathways. Thus, the enzymes for the b-oxidation of fatty acids are found in peroxisomes. The importance of this phenomenon in toxicity and especially carcinogenicity is discussed later in this chapter. The types of compounds, which cause the proliferation, are generally acids or compounds, which can be metabolized to acids. Thus, the hypolipidemic drug clofibrate, and a number of similarly acting drugs, will cause the proliferation. Phthalate esters, which are commonly used as plasticizers, are another group of compounds, which have been shown to be active. The results of exposure in rodents are the following: a large increase in the numbers of peroxisomes (e.g., 140%); a large increase in liver weight (e.g., 3.9–8.5%body weight); an increase in DNA content (e.g., 1.5–2); increases in RNA and protein synthesis; large increases in the enzymes of b-oxidation such as palmitoyl CoA oxidase (e.g., 6–15, butsome enzymes maybe increased up to 150); and increases in the cytochrome P-450 enzymes,which metabolize fatty acids such as lauric acid (CYP4A1, e.g., 5–10). This type of response is achieved after exposure to a compound such as clofibrate for 28 days. There is clearly a modulation of gene expression occurring for RNA, DNA, and protein synthesis to be increased, and this seems to be mediated by a receptor interaction. Several possible mechanisms have been proposed to account for the phenomenon, and these are not necessarily exclusive: (i) interaction between peroxisome proliferators and a receptor[peroxisome proliferator–activated receptor (PPAR)]; (ii) perturbation by peroxisome proliferators of lipid metabolism, leading to substrate overload; (iii) action of peroxisomal proliferators as substrates for lipid metabolism. Thus, there is evidence for a receptor, which can be activated by peroxisome proliferators in vitro. This interaction seems to activate genes involved with peroxisomal and microsomal fatty acid oxidation. However a perturbation, such as inhibition, of lipid metabolism may also be involved and could lead to increased levels of an endogenous ligand for the receptor. For example, fatty acids such as oleic acid are known to bind to and activate the PPAR in vitro. If drugs and other chemicals, which are peroxisomal proliferators, acted as substrates for enzymes, such as those catalysing boxidation, they could perturb lipid metabolism, leading to changes in the levels of crucial fatty acids. Therefore, increased peroxisomal enzyme activity could be response. an adaptive The receptor protein (52 kDa) is a member of the steroid hormone receptor superfamily, which has a DNA-binding as well as ligand-binding domain. Another receptor, the retinoid X receptor is also involved, and after binding of the peroxisome proliferator, the two receptors form a heterodimer. This binds to a regulatory DNA sequence known as the peroxisome proliferator response element. The end result of the interaction between peroxisome proliferators and this system is that genes are switched on, leading to increases in synthesis(induction) of both microsomal and peroxisomal enzymes and possibly hyperplasia. Structure activity studies in vitro have revealed that the relative potency of peroxisome proliferators seems to be determined by a combination of lipophilicity and the calculated binding affinity to the mouse PPARa ligand-binding domain. The mechanisms underlying some of these types of injury will be discussed in general terms below and in chapter 7. Detection of Hepatic Damage Simple quantitative tests can be used such as measurement of the liver weight/body weight ratio. Overt damage to the liver can be detected by light and electron microscopy of liver sections. However, damage can be detected by other noninvasive means such as the urinaryexcretion of conjugated bilirubin or the amino acid taurine.Various parameters may be measured in plasma. Thus, determination of the enzymesaspartate transaminase (AST) and alanine transaminase (ALT) is the most common means of detecting liver damage, the enzymes being raised several fold in the first 24 hours after damage. However, there are a number of other enzymes, which may be used as markers. Plasma bilirubin can also be measured, being increased in liver damage, and plasma albuminis decreased by liver damage (although also by renal damage). Liver function may bedetermined using the hepatic clearance of a dye such as sulfobromophthalein. 6.2.4 Kidney 6.2.5 Lung 6.2.6 Other Target Organs 6.3 MECHANISM AND RESPONSE IN CELLULAR TOXICITY 6.3.1 Primary Events 6.3.2 Secondary Events 6.3.3 Tertiary Events 6.3.4 Protective Mechanisms 6.4 PHARMACOLOGICAL, PHYSIOLOGICAL, AND BIOCHEMICAL EFFECTS 6.4.1 Anoxia 6.4.2 Inhibition of Cellular Respiration 6.4.3 Respiratory Failure 6.4.4 Disturbances of the CNS 6.4.5 Hyper-/Hypotension 6.4.6 Hyper-/Hypoglycemia Toxic Responses to Foreign Compounds 235 6.4.7 Anesthesia 6.4.8 Changes in Water and Electrolyte Balance 6.4.9 Ion Transport 6.4.10 Failure of Energy Supply 6.4.11 Changes in Muscle Contraction/Relaxation 6.4.12 Hypo-/Hyperthermia 6.4.13 Heightened Sensitivity 6.5 DEVELOPMENTAL TOXICOLOGY— TERATOGENESIS 6.5.1 Introduction, 6.5.2 Characteristics of Teratogenesis 6.5.3 Mechanisms of Teratogenesis 6.5.4 Role of Metabolic Activation 6.5.5 Transplacental Carcinogenesis 6.6 IMMUNOTOXICITY 6.6.1 Immunosuppression 6.6.2 Immunoenhancement 6.6.3 Hypersensitivity 6.6.4 Characteristics of Immunological Reactions 6.7 GENETIC TOXICITY 6.7.1 Introduction 6.7.2 Mutagenesis 6.7.3 Direct Interaction with DNA 6.7.4 Primary DNA Damage 6.7.5 Gene Mutations 6.7.6 Base Substitutions 6.7.7 Frameshift Mutations 6.7.8 Clastogenesis 6.7.9 Aneugenesis 6.7.10 DNA Repair 6.7.11 Mutagenesis in Mammals 6.7.12 Determination of Mutagenicity and its Relation to Carcinogenicity 6.8 CHEMICAL CARCINOGENESIS 6.8.1 Introduction 6.8.2 Mechanisms Underlying Carcinogenesis 6.8.3 Initiation 6.8.4 Promotion 6.8.5 Progression 6.8.6 DNA Repair 6.8.7 Non-Genotoxic Mechanisms 6.8.8 Cell Proliferation 6.8.9 Testing for Carcinogenicity and Relation of Mutagenicity