Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
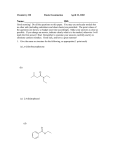
A missense mutation in Caenorhabditis elegans prohibitin 2 confers an atypical multidrug resistance Iryna Zubovych, Thomas Doundoulakis, Patrick G. Harran, and Michael G. Roth* Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 75390-9038 Communicated by Xiaodong Wang, University of Texas Southwestern Medical Center, Dallas, TX, August 24, 2006 (received for review July 1, 2006) Me Me O Me N CO2H N H NHMe N Me O Pr-i Me 1 Me Me O Bu-t Me N CO2H N H NHMe R I O Pr-i Me 2 R = H (HTI-286) 3 R = Br 4 R=A antimitotic 兩 hemiasterlin dentifying the molecular target(s) and mechanisms of biological activity of small molecules remains a major challenge. Among the tactics available, the techniques of classical forward genetics are particularly attractive. A genetic screen or selection for organisms resistant to a drug requires no assumptions about the drug target, mechanism of action, or biological pathway. Such a screen can also reveal mechanisms that can inactivate a compound or suppress its effects, issues of great importance for the development of effective therapeutics. We chose the roundworm Caenorhabditis elegans as a genetic model system to investigate possible pathways involved in resistance to the potent antimitotic hemiasterlin. Hemiasterlin is one of the naturally occurring toxins found to act on tubulin (1). Hemiasterlin poisons spindle assembly in cultured cells, leading to growth arrest in mitosis (2). Among hemiasterlin analogs evaluated, HTI-286 (compound 2, Fig. 1) has been characterized most extensively. This compound inhibits microtubule assembly in vitro (3), and mutations in ␣- or -tubulin that increase microtubule stability confer resistance to HTI-286 in ovarian carcinoma cells (4). Because HTI-286 is in clinical development as a promising cancer chemotherapeutic (5), we initiated genetic experiments to identify additional pathways associated with hemiasterlin toxicity. We have isolated eight mutants resistant to a functional analog of hemiasterlin and characterized the mutated gene in one recessive mutant as that encoding prohibitin 2. We show that a single amino acid change in prohibitin 2 protects C. elegans not only from hemiasterlin but also from several other toxins that bind tubulin and from camptothecin, an inhibitor of DNA replication. However, this mutation confers no resistance to the tubulin polymerization inhibitor nocodazole or the actin stabilizer phalloidin. Bu-t Me Me O Bu-t Me CO2H N N H NHMe R O Pr-i 5 R=A O A= HN NH H N S O Fig. 1. Hemiasterlin (1), HTI-286 (2), and synthetic variants (3–5) used in this study. Compounds 3 and 4 were designed and synthesized based on earlier structure兾activity relationships established by Nieman et al. (7). Compound 5 is an inactive control. (Table 1, which is published as supporting information on the PNAS web site). Saturated compound 5, as anticipated by precedent (7), was found to be inactive and served as a structurally related, biotinylated negative control. Compound 3 inhibited microtubule formation in vitro (Fig. 5, which is published Results Compound 3 Is Cytotoxic, Binds Tubulin, and Inhibits Tubulin Polymerization. We synthesized three variants of hemiasterlin (Fig. 1) and compared their properties to those reported for natural hemiasterlin. Compound 3 intoxicated cells at concentrations similar to those reported for hemiasterlin (6, 7), and the biotinconjugate of compound 3, compound 4, was similarly toxic www.pnas.org兾cgi兾doi兾10.1073兾pnas.0607338103 Author contributions: I.Z., P.G.H., and M.G.R. designed research; I.Z. performed research; T.D. and P.G.H. contributed new reagents兾analytic tools; I.Z., P.G.H., and M.G.R. analyzed data; and I.Z., P.G.H., and M.G.R. wrote the paper. Conflict of interest statement: X.W. is part of the program grant that produced this work. *To whom correspondence should be addressed. E-mail: [email protected]. © 2006 by The National Academy of Sciences of the USA PNAS 兩 October 17, 2006 兩 vol. 103 兩 no. 42 兩 15523–15528 GENETICS Hemiasterlin is a potent antimitotic peptide that interferes with microtubule dynamics at picomolar concentrations in cell culture. The molecule largely eludes P glycoprotein-mediated drug efflux, and an analog is currently being evaluated in clinical trials as cancer chemotherapy. From a nonclonal genetic screen in Caenorhabditis elegans we isolated eight independent mutants resistant to a synthetic hemiasterlin analog. In one recessive mutant, phb2(ad2154), a point mutation in prohibitin 2 (E130K) protects worms from drug-induced injury. Data indicate that direct binding of hemiasterlin to prohibitin 2 is unlikely. In fact, C. elegans phb2(ad2154) was also found to be resistant to numerous other drugs that bind tubulin and to camptothecin, yet this mutant was sensitive to nocodazole and phalloidin. Thus, prohibitin 2 is implicated in a previously uncharacterized pathway of multidrug resistance. Fig. 2. Compound 4 binds tubulin but not PHB2. (A) Toxic 4 binds tubulin and nontoxic 5 does not. Preparations of purified tubulin, tubulin enriched in microtubule-associated proteins, and a HeLa cell lysate were incubated overnight with 5 M biotinylated hemiasterlin probes or an equal volume of the DMSO vehicle and then precipitated with neutravidin beads. The precipitates were analyzed by PAGE, and tubulin was detected by immunoblotting. A result typical of five experiments is shown. (B) Compound 4 fails to pull down recombinant PHB2. Purified recombinant PHB2 was prepared as described and subjected to a precipitation experiment with biotinylated compound 4 as described for tubulin in Materials and Methods. Samples were resolved by PAGE, and PHB2 was detected by immunoblotting. Lane 1, purified PHB2 from the starting material; lane 2, the neutravidin biotin-binding matrix alone; lane 3, PHB2 incubated with DMSO and biotin-binding matrix; lane 4, PHB2 incubated with 5 M compound 4 and biotin-binding matrix; lane 5, PHB2 incubated with 5 M compound 5 and biotin-binding matrix. as supporting information on the PNAS web site), and cells overexpressing MDR1 displayed only 3-fold resistance to compound 3 but were 150-fold resistant to taxol and peloruside A (data not shown), in agreement with a report that HTI-286 is a poor substrate for the P glycoprotein pump (3). Cytotoxic, biotinylated compound 4, but not the control biotinylated compound 5, was able to specifically affinity-purify tubulin (Fig. 2A) from HeLa cell lysate. Thus, compound 3 retained the functional properties of the natural product and was an appropriate probe of hemiasterlin biology. Compound 3 Is Highly Toxic for C. elegans. To determine the effect of compound 3 on C. elegans, we examined development and health of wild-type L1 larvae growing in liquid culture at various concentrations of compound. Vehicle (DMSO) control had no effect on worms. The lowest toxic dose of compound 3, 700 nM, produced normal sized, but paralyzed, animals. Larvae growing at 1 M compound 3 developed into fertile, dumpy adults (Fig. 3; and see Movies 1 and 2, which are published as supporting information on the PNAS web site). At 2 M, the dumpy phenotype became more severe, and animals laid eggs that never developed (embryonic lethality). All larvae incubated with 5 M compound 3 were paralyzed within 12 h, growth-arrested at the L1 stage, and died within 2– 4 days. The significantly higher concentration of compound 3 required to observe a phenotype in C. elegans compared with mamma15524 兩 www.pnas.org兾cgi兾doi兾10.1073兾pnas.0607338103 Fig. 3. When grown in 1 M compound 3, wild-type worms and phb2(ad2154) mutants differ in morphology. (A) Wild-type worms grown for 4 days in normal culture conditions. (B) Wild-type animals grown for 4 days in 1 M compound 3 become short, paralyzed, and morphologically abnormal but lay eggs and reproduce (middle two animals). (C) Phb-2(ad2154) animals grown for 4 days in 1 M compound 3 develop into healthy adults. Animals to the right are laying eggs. lian cells was also observed for other drugs tested (Fig. 4). Some of the drugs tested, such as colchicines, cytochalasin B, or dolastatin 15, were not toxic for worms at all. Characteristics of Mutants Resistant to Compound 3. We next per- formed a genetic screen for mutants that were resistant to compound 3. After mutagenesis with ethyl methanesulfonate, we isolated eight fully penetrant mutants: two dominant and six recessive. We found 1 M compound 3 to be the most convenient concentration to assay, because adults reproduced and offspring of a single animal could be characterized as either sensitive (paralyzed fertile dumpy) or resistant (healthy animals). Two animals with strong phenotypes were outcrossed three times and characterized. One carried a dominant mutation (ad2155dm) that was mapped to a region on the left arm of chromosome III, between cosmids C32A3 and W03A5. The only tubulin gene (tbb-2) located in this area was sequenced and found to be wild type. The other animal, phb2(ad2154) carried a recessive mutation and appeared unaffected at 1 M compound 3 (see Movies 3 and 4, which are published as supporting information on the PNAS web site). The mutation conferring resistance in phb-2(ad2154) was identified by standard genetic methods and DNA sequencing, as described in Materials and Methods. Injecting a PCR fragment containing the wild-type phb-2 gene with 1 kb of Zubovych et al. upstream and 369 bp of downstream sequence into phb2(ad2154) worms resensitized them to the drug, confirming that the mutation in phb-2 was responsible for drug resistance. This mutation was a G-to-A transversion, resulting in a change of an evolutionarily conserved glutamic acid to lysine at amino acid 130 (Fig. 6, which is published as supporting information on the PNAS web site). mnDf30 and mnDf96 worms bear a deletion covering the phb-2 gene. Crossing phb-2(ad2154) males with these deficient strains produced resistant progeny in 1 M compound 3 that were resistant to the same drug concentrations as their phb-2(ad2154) Zubovych et al. parent. Thus, a single copy of the phb-2 gene conferred drug resistance. Mating phb-2(ad2154) males with each of the other recessive mutants isolated in our screen produced drug-sensitive progeny. DNA sequencing revealed that the remaining mutants were wild type for phb-2 and phb-1. The phenotype reported for worms in which phb-2 was targeted by RNAi was similar to that observed in wild-type worms treated with compound 3 (8). To determine whether PHB2 was a direct target of compound 3, we incubated the biotinylated compound 4 with pure recombinant PHB2. Under conditions where compound 4 was able to affinity-purify PNAS 兩 October 17, 2006 兩 vol. 103 兩 no. 42 兩 15525 GENETICS Fig. 4. phb-2(ad2154) worms are resistant to many toxins but not to nocodazole or phalloidin. Approximately 300 wild-type N2 (squares) or phb-2(ad2154) (triangles) L1 larvae were incubated in drugs at indicated concentrations for 4 days, and the healthy, morphologically normal, mobile adult worms with eggs were counted. For each concentration, three wells were scored in an experiment. Average values were graphed ⫾ SEM. Similar results were obtained in at least five independent experiments. tubulin in vitro (Fig. 2 A), we could detect no interaction with PHB2 (Fig. 2B). Phb-2(ad2154) Worms Are Resistant to Other Drugs. We next determined the sensitivity of N2 worms to other tubulin-binding toxins, both microtubule stabilizers and destabilizers (Fig. 4) by treating L1 larvae with the drugs and scoring their ability to develop normally. Interestingly, compound 3 and nocodazole exhibited visually similar effects on wild-type C. elegans, causing them to develop into paralyzed, dumpy worms (Fig. 3B). However, nocodazole, unlike compound 3, did not affect fertility and viability; dumpy animals survived and reproduced at concentrations as high as 200 M. Cryptophycin-1 acted similarly to compound 3, except that adults were mobile. All other tested drugs arrested worm development at larval stages, but animals escaping this arrest grew to healthy, fertile adults. Thus, compounds having comparable inhibitory actions on tubulin and mitosis affected C. elegans development distinctly. To determine whether phb-2(ad2154) conferred resistance to other drugs, we counted the number of N2 and phb-2(ad2154) worms grown in the presence of compounds that grew to be healthy gravid adults (Fig. 4). phb-2(ad2154) worms were resistant to compound 3 but not to nocodazole. phb-2(ad2154) animals showed similar levels of resistance to compound 3, dolastatin 10, cryptophycin-1, paclitaxel, peloruside A, vincristine, vinblastine, and phomopsin A (Fig. 4). Because certain of these drugs are excellent substrates for the P glycoprotein efflux pump and compound 3 is not, we conclude that this mechanism of resistance is not contributing to the phenotype. We also tested toxic compounds that do not target tubulin, camptothecin (a DNA topoisomerase I inhibitor) and phalloidin (stabilizes actin). The phb-2(ad2154) mutant was resistant to camptothecin and fully sensitive to phalloidin, indicating that mutant PHB2 protects worms from drug-induced toxicity specifically. Importantly, the dominant mutant (ad2155dm) displayed similar drug responses. ad2155dm was fully sensitive to nocodazole and phalloidin and cross-resistant to the remaining compounds (data not shown), indicating that these two mutants are likely to be on the same pathway. Discussion Determining the target of bioactive compounds and their mechanisms of action is a challenge and requires the combined use of organic synthesis, biochemistry, and genetics. Even when a target of a compound has been previously identified, genetics has the potential to reveal unsuspected aspects of the biological response and novel mechanisms of resistance. In the case of our derivatives of hemiasterlin, we have mapped resistance to multiple toxins to PHB2. Several very different functions have been proposed for PHB2, none of which directly explain the drug resistance we observe in phb-2(ad2154). PHB2 (also called BAP37 or REA) is a ubiquitously expressed, evolutionary conserved protein with high similarity to prohibitin 1 (PHB1, BAP32). PHB1 and PHB2 form a high-molecular-weight complex and may function together in the mitochondria (9, 10). Prohibitins have been reported to be localized to the plasma membrane and the nucleus and in mitochondria with the N terminus anchored into the inner mitochondrial membrane and C terminus protruding into the intermembrane space (9, 11–13). Prohibitins have been suggested to have multiple cellular functions, including cell proliferation, senescence and aging, tumor suppression, mitochondrial protein folding, and transcriptional repression. PHB1 was overexpressed in certain carcinomas (14–16). Although called a ‘‘tumor suppressor,’’ how PHB1 participates in carcinogenesis is unclear. Genome-wide transcriptional profiling in C. elegans found phb-2 (T24H7.1) to be expressed at all developmental stages from egg to adult, with the highest prevalence in embryo and larvae and lowest in 2-week-old worms (17). Depending on the 15526 兩 www.pnas.org兾cgi兾doi兾10.1073兾pnas.0607338103 degree to which protein expression was reduced, phb-2 RNAi or phb-1 RNAi worms exhibited embryonic lethality, reduced body size, and developmental-delay phenotypes (8). Interestingly, partial down-regulation of prohibitin expression in petunia resulted in dwarf plants. Cells in the silenced dwarf flowers were larger in diameter but significantly reduced in number, suggesting an effect on cell proliferation (18). Homozygous PHB2knockout mouse embryos die in early development, whereas heterozygous animals survive and can reproduce (19). Our results indicate that a single amino acid change in PHB2 protects C. elegans from some but not all drug effects, because phb-2(ad2154) animals are fully sensitive to nocodazole and phalloidin. The phb-2 RNAi phenotype in C. elegans is dumpy and embryonic lethal, very similar to the phenotype of wild-type worms grown in compound 3. We could not detect the binding of biotinylated compound 4 to PHB2, and multiple drug resistance also suggests that PHB2 is not a direct target of compound 3. If PHB2 acts downstream of certain toxins in a mechanism responsible for cell death, and phb-2(ad2154) is defective in this response, not all tubulin-binding antimitotics induce cell death through a common pathway. Alternatively, PHB2 might function to control the intracellular concentration of certain toxins. If the recessive mutation in phb-2(ad2154) is a partial loss or change in function, PHB2 might inhibit drug efflux or degradation mechanisms or activate drug influx. It is not known how the drugs used in this study penetrate worms. We have not compared the drug concentrations inside wild-type worms and phb-2(ad2154) worms, because the differences in effective drug concentrations are small, and drugs in the mutant may be retained in the intestinal lumen or excreted into pseudocoelomic fluid. Further analysis of other mutants, particularly the (ad2155dm) mutant that shows a similar response, will provide a more comprehensive view of this pathway. We believe that finding the molecular mechanisms involved may have clinical implications to combat multidrug resistance. Materials and Methods Synthetic Reagents. Hemiasterlin analog compound 3 was synthesized according to published protocols (7). The route used was unchanged, except that it was initiated with  -(3bromophenyl) isovaleric acid instead of -phenylisovaleric acid. We synthesized compound 3 instead of compound 2 because compound 3 was the precursor to biotinylated probe compound 4, which was prepared by coupling a protected form of compound 3 (see supporting information) with BocHN(CH2)6CCH [2 mol% (t-Bu3P)2Pd, 3 mol% CuI, (i-Pr)2NEt, p-dioxane, rt]. The resulting product was saponified (LiOH, aq MeOH), exposed to excess trifluoroacetic acid (CH2Cl2, rt), and treated with the N-hydroxysuccinimidyl ester of L-biotin (Sigma, St. Louis, MO). Inactive control compound 5 was synthesized in a directly analogous manner. Both reagents were purified to homogeneity by preparative HPLC (5 M C18, CH3CN兾H2O gradient) and possessed analytical data in full accord with their proposed structures. Cell Lines, Reagents, and Cytotoxicity. All cell lines tested were purchased from American Type Culture Collection (Manassas, VA). SK-MEL-5 cells were grown in modified Eagle’s medium (MEM; Invitrogen, Carlsbad, CA) with 1.5 g兾liter NaHCO3 supplemented with 10% FBS (GIBCO, Carlsbad, CA) and 1 mM sodium pyruvate (Invitrogen). BSC-1 and MCF-7 cells were maintained in MEM with 3.5 g兾liter NaHCO3 and 10% FBS, CHO in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 5% FBS, 10 mM Hepes, and 2 mM L-proline. HeLa cells were cultured in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, and 10 mM Hepes. All culture media contained 1% of penicillin–streptomycin solution (Invitrogen). To characterize the antiproliferative effect of drugs, 30,000 cells Zubovych et al. Tubulin and in Vitro Polymerization Assays. MAP-enriched tubulin was purified from four fresh bovine brains (Dallas City Packing, Dallas, TX) by using a protocol from the Mitchison laboratory with slight modifications. MAP-enriched tubulin aliquots were stored in a ⫺80°C freezer until use. Pure tubulin was purchased from Cytoskeleton (Denver, CO). Binding to Biotinylated Compounds. After thawing, tubulin aliquots were centrifuged at 14,000 ⫻ g for 10 min at 4°C to remove aggregates, and then the supernatant was diluted to 0.5 mg兾ml final concentration in PEM buffer containing 1% Nonidet P-40 (Calbiochem, San Diego, CA). Compounds 4 or 5 were added at 5 M in a total reaction volume of 200 l in 0.5-ml tubes and incubated at 4°C overnight on a rotator. Fifty microliters of Immobilized Neutravidin Biotin Binding Protein beads (Pierce, Rockford, IL) was added to each tube for 30 min, the reactions were carefully transferred to 1.5-ml Eppendorf tubes and washed in incubation buffer seven times at 4°C, and precipitates were analyzed by SDS兾PAGE. After electrophoresis, proteins were electrotransferred to 0.45-m nitrocellulose membrane (Protran BA85), immunoblotted with a 1:3,000 dilution of monoclonal anti-␣-tubulin antibody (Sigma), followed by a 1:3,000 dilution of HRP-conjugated goat anti-mouse (Bio-Rad, Hercules, CA), and then detected with ECL reagent (PerkinElmer Life Sciences, Boston, MA) according to the manufacturer’s instructions. To prepare HeLa cell lysate, cells were lysed directly on 15-cm tissue culture plate by adding 4 ml of PEM buffer supplemented with 1% Nonidet P-40 and protease-inhibitor mixture (Roche, Indianapolis, IN). The cell lysate was incubated for 1 h on a rotator at 4°C and then centrifuged at 14,000 ⫻ g for 40 min at 4°C. The supernatant was collected and incubated with biotinconjugated compounds. The ability of the compounds to pull down tubulin from the HeLa lysate was determined as for purified tubulin. Worm Culture. Worms were grown at 20°C by using standard culture conditions (20) with slight modifications (21). Wild-type C. elegans var. Bristol N2 was the parent of all mutant strains. For genetic mapping experiments, strains used were Hawaiian strain CB4856; CB768 bli-2 (e768) II; CB187 rol-6 (e187) II; SP543(mnDf30) unc-4(e120)兾mnC1 dpy-10(e128) unc-52(e444) II; and SP788 unc-4(e120) mnDf96兾mnC1 dpy-10(e128) unc52(e444) II. Worms were fed bacterial strain HB101 (22). To obtain first-stage synchronous larvae (L1s), eggs were purified by alkaline hypochlorite treatment (23), rinsed five times in M9 buffer (24), suspended in 4 ml of M9, and placed on a shaker for 13–16 h. Drug tests were performed in 150 l of M9 buffer containing 5 mg兾liter cholesterol, HB101, and compounds at room temperature (⬇22°C) in 48-well plates (FALCON; Becton Dickinson, Franklin Lakes, NJ). Drugs. Paclitaxel (taxol), nocodazole, vinblastine, vincristine, camptothecin, phomopsin A, and phalloidin are commercially available (Sigma). Dolastatin 10 was a kind gift from Gregory Kalemkerian (University of Michigan Medical Center, Ann Arbor, MI), cryptophycin 1 was generously provided by Richard Himes and Gunda Georg (University of Kansas, Lawrence, KS), and peloruside A was obtained from Jef De Brabander (University of Texas Southwestern Medical Center). Stock solutions were prepared in DMSO, aliquoted, and stored at ⫺80°C. Working dilutions were prepared in M9 medium immediately before use. Zubovych et al. Genetic Screen for Compound 3-Resistant Mutants. Late L4 N2 animals were mutagenized by exposure to 47 mM ethyl methanesulfonate for 4 h. Nine-hundred thousand L1 F2 progeny of mutagenized parental animals were plated in 4 M compound 3, and the worms were scored for body shape and ability to move. The progeny of candidates were examined for the same criteria, including the ability to reproduce in the presence of the drug. Eight animals had a mutant phenotype. Sequencing. We amplified all 65.7 kb of genomic DNA from the phb-2(ad2154) mutant from snp uCE2–1658 to snp㛭C29F5[1] and sequenced the PCR products. We identified the only mutation present in that region in the T24H7.1 (phb-2) gene. Complementation Experiments. Cosmids were prepared by using the Plasmid Isolation Mini kit (Qiagen, Valencia, CA) and injected, by using standard injection techniques, into phb2(ad2154) at a concentration 20 ng兾l with rol-6 DNA as a marker for transformation at a concentration 50 ng兾l (25). Genetic Mapping of phb-2(ad2154) and Rescue Experiments. By standard polymorphism mapping, a recessive drug-resistance mutation was linked to chromosome II. More precise mapping was performed by flanking the mutation between bli-2(e768) and rol-6(e187) visible markers. Briefly, drug-resistant mutant males were mated with bli-2 hermaphrodites, and drug-resistant bli-2 males were selected and mated with rol-6 hermaphrodites to obtain drug-resistant triple-mutant animals. CB4856 males were mated with triple mutants and, from the F2 generation, recombinant Bli-nonRol worms were selected, made homozygous, and tested for the compound 3-sensitive or -resistant genotype. After SNP analysis of isolated recombinants, the mutation was mapped between snp uCE2-1658 and snp㛭C29F5[1]. Mating with two strains, mnDf30 and mnDf96, each having a deficiency spanning the region, confirmed the location of the mutation. This 65-kb region was covered by three overlapping clones, T24H7, F13H8, and C29F5, and included 19 genes. We sequenced this region simultaneously with rescue experiments. Cosmid T24H7 rescued the mutant phenotype, and sequence data identified only a single mutation present in the 65.7 kb of DNA in the T24H7.1 gene. For rescue with a genomic fragment, a 2807-bp PCR product spanning the phb-2 locus with 1 kb of upstream sequence and 369 bp of downstream sequence was amplified from T24H7 cosmid DNA and injected into phb-2 (ad2154) mutants at a concentration 0.5 ng兾l with rol-6 (50 ng兾l). The following primers were used for amplification: forward, CGTGAAGTAGATCGATTCATG; reverse, CTGCATGCTGTATTGATCGA. We isolated 13 independent stable transgenic lines, and 12 of them were converted to drug sensitivity by the presence of wild-type phb-2; 80–100% transgenic worms of each line displayed dumpy phenotype in the presence of 1 M compound 3. Drug Toxicity Experiments with Worms. Escherichia coli HB101 from cultures grown overnight in 1 liter of LB broth supplemented with 0.05 g兾liter streptomycin were washed twice with water, resuspended in 150 ml of M9 buffer supplemented with cholesterol (5 mg兾liter), and dispensed into 48-well tissue culture plates to provide food for worms. Compounds were added to the wells to the desired concentrations. DMSO was always loaded as a control, and DMSO concentrations were kept below 1.5%. Approximately 300 synchronized L1 larvae were plated into each well of 48-well tissue culture plates. After 3–4 days, when all animals grown in DMSO had become adults, the content of each well was transferred to an empty Petri dish, and the number of healthy adult worms was counted. Photography. Images of worms were taken with a MaxCam CM10 digital camera (Finger Lakes Instrumentation, Lima, NY) on an PNAS 兩 October 17, 2006 兩 vol. 103 兩 no. 42 兩 15527 GENETICS were plated per well in 12-well tissue culture plates. The next day, cells were treated with varying concentrations of compound, and cell numbers were determined for 4 days by using a Beckman Cell Counter (Coulter, Miami, FL). Axiophot microscope (Zeiss, Oberkochen, Germany) using Nomarsky optics and a 5⫻ objective. Supporting movies were recorded with a KP-160 camera (Hitachi, Tokyo, Japan) at a rate of 30 frames per second and digitized by using a Matrox RTX-10 video capture board (Matrox Electronic Systems, Dorval, QC, Canada). PHB2 Expression, Purification, and Pull-Down Experiments. PHB2 CA). The washed beads were incubated overnight with thrombin (Amersham) at room temperature in 1⫻ PBS containing 2.5 mM CaCl2, and then thrombin was inactivated with 1 mM PMSF. The released PHB2 (0.2 mg兾ml in PBS, with 1% Nonidet P-40 added) was incubated with biotinylated compounds and neutravidin beads and analyzed as described for experiments with tubulin, except that the nitrocellulose membrane was probed with a 1:500 dilution of PHB2 antibody (BioLegend, San Diego, CA). cDNA in a pCMV6-XL5 vector was purchased from OriGene Technologies (Rockville, MD). The cDNA was amplified by PCR using the following primers: forward, containing an EcoRI restriction site, GATCAGAATTCTAATGGCCCAGAACTTGAAG, and reverse, containing an XhoI restriction site, GATCACTCGAGTCATTTCTTACCCTTGATGA, and inserted into the pGEX-KG GST-fusion vector. After DNA sequencing to verify the GST-PHB2 fusion sequence, the plasmid was transformed into BL21-CodonPlus-RIL cells (Stratagene, La Jolla, CA). Protein expression was induced with IPTG for 4 h at room temperature, and the GST-PHB2 from a cell lysate was bound to glutathione Sepharose (Amersham, San Francisco, We thank Leon Avery (University of Texas Southwestern Medical Center) for sharing strains and reagents and for discussion, Anthony Burgett for the tubulin purification, Audrey Fraser (Wellcome Trust Sanger Institute, Hinxton, Cambridge, U.K.) for cosmid clones, Boris Shtonda for help with photography, Gregory Kalemkerian for dolastatin 10, Gunda Georg and Richard Himes for providing cryptophycin-1, and Jef De Brabander for peloruside A. Some strains used in this work were supplied by the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN), which is funded by the National Institutes of Health National Center for Research Resources. This work was supported by National Cancer Institute Grant P01CA95471. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant C06 RR-15437. 1. Janin YL (2003) Amino Acids 25:1–40. 2. Anderson HJ, Coleman JE, Andersen RJ, Roberge M (1997) Cancer Chemother Pharmacol 39:223–226. 3. Loganzo F, Discafani CM, Annable T, Beyer C, Musto S, Hari M, Tan X, Hardy C, Hernandez R, Baxter M, et al. (2003) Cancer Res 63:1838–1845. 4. Poruchynsky MS, Kim JH, Nogales E, Annable T, Loganzo F, Greenberger LM, Sackett DL, Fojo T (2004) Biochemistry 43:13944–13954. 5. Ayral-Kaloustian S, Zask A (2005) Drugs Future 30:254–260. 6. Gamble WR, Durso NA, Fuller RW, Westergaard CK, Johnson TR, Sackett DL, Hamel E, Cardellina JH, II, Boyd MR (1999) Bioorg Med Chem 7:1611– 1615. 7. Nieman JA, Coleman JE, Wallace DJ, Piers E, Lim LY, Roberge M, Andersen RJ (2003) J Nat Prod 66:183–199. 8. Artal-Sanz M, Tsang WY, Willems EM, Grivell LA, Lemire BD, van der Spek H, Nijtmans LG (2003) J Biol Chem 278:32091–32099. 9. Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA (2000) EMBO J 19:2444–2451. 10. Tatsuta T, Model K, Langer T (2005) Mol Biol Cell 16:248–259. 11. Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S (2003) J Biol Chem 278:47853–47861. 12. Ikonen E, Fiedler K, Parton RG, Simons K (1995) FEBS Lett 358:273–277. 13. Terashima M, Kim KM, Adachi T, Nielsen PJ, Reth M, Kohler G, Lamers MC (1994) EMBO J 13:3782–3792. 14. Asamoto M, Cohen SM (1994) Cancer Lett 83:201–207. 15. Srisomsap C, Subhasitanont P, Otto A, Mueller EC, Punyarit P, WittmannLiebold B, Svasti J (2002) Proteomics 2:706–712. 16. Wang KJ, Wang RT, Zhang JZ (2004) World J Gastroenterol 10:2179–2183. 17. Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL (2000) Science 290:809–812. 18. Chen JC, Jiang CZ, Reid MS (2005) Plant J 44:16–24. 19. Park SE, Xu J, Frolova A, Liao L, O’Malley BW, Katzenellenbogen BS (2005) Mol Cell Biol 25:1989–1999. 20. Sulston JE, Hodgkin JA (1988) Methods, the Nematode Caenorhabditis elegans (Cold Spring Harbor Lab Press, Cold Spring Harbor, NY). 21. Avery L (1993) Genetics 133:897–917. 22. Boyer HW, Roulland-Dussoix D (1969) J Mol Biol 41:459–472. 23. Emmons SW, Klass MR, Hirsh D (1979) Proc Natl Acad Sci USA 76:1333–1337. 24. Brenner S (1974) Genetics 77:71–94. 25. Mello C, Fire A (1995) Methods Cell Biol 48:451–482. 15528 兩 www.pnas.org兾cgi兾doi兾10.1073兾pnas.0607338103 Zubovych et al. SupplFig1.pzf:Layout 1 - Thu Jun 1 11:18:09 2006 Tubulin Polymerization absorbance 0.5 Peloruside A Taxol 0.4 DMSO 3 0.3 500 1000 seconds Supplementary Figure 1 1500 2000 YEERVLPSIVNEVLKSVVAKFN YEERVLPSIVNEVLKSVVAKFN YDERVLPSIVNEVLKSVVAKFN WEERVLPSICNEVLKGVVAKFN YDERVLPSIVNEVLKAVVAQFN Figure 5 - human rat zebrafish C. elegans yeast