Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
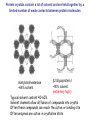
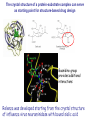
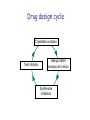
Crystallography: An introduction Harma Brondijk Crystal and Structural chemistry, Utrecht University Crystallography Phase problem Construct Pure protein Crystal 3D structure X-ray Diffraction Electron density Why X-ray crystallography? (Light) microscope: Limitations: Object needs to be larger than the wavelength of the light (visible light 400-700 nm, atoms = 0.15 nm apart) X-rays(0.08-0.6 nm) cannot be focussed by lenses Molecules are very weak scatterers X-ray diffraction of a crystal A crystal contains many molecules in identical orientation Diffracted x-rays of individual molecules ‘add up’ (positive interference) to produce strong reflections Computers can simulate a lens and reconstruc the image (Fourier transform) Growing crystals Hanging drop: 1 μl protein solution+ 1 μl reservoir solution Reservoir: precipitant solution eg. 1 M NaCl or 30% PEG-4k [precipitant] and [protein] slowly rise as drop equilibrates with reservoir [protein] precipitation Nucleation & growth growth Soluble protein [precipitant] Getting your data X-ray data are measured on frozen crystals (~100K) Frozen crystal mounted in loop for Xray data collection In house X-ray data collection set up For high quality X-ray data collection extremely intense synchrotron beam lines - like here in Grenoble are used Fourier transform (phase problem) Raw data: Thousands of intensities of reflections Electron density Each diffraction spot (reflection) contains information on the position of every atom! The degree of order in the crystal determines the quality of the diffraction data and ultimately the quality of the final atomic model “low resolution” “high resolution” The precision of the atomic model is mainly determined by the maximal resolution to which the crystal diffracts X-rays d=4Å d=3Å d=2Å d=1Å Atomic resolution Some real life examples 1.8 Å structure: core vs surface loop at 2σ 3.1 Å resolution, well ordered core of the protein What’s a crystal structure? Different representations of an Fab fragment of monoclonal antibody 82D6A3 bound to the collagen binding domain of human von Willebrand factor Things you - as a potential user of crystallographic data - should know about crystals and crystal structures Protein crystals contain a lot of solvent and are held together by a limited number of weak contacts between protein molecules Acetylcholinesterase ~68% solvent 2 Glycoprotein I ~90% solvent (extremely high!) Typical solvent content 40-60% Solvent channels allow diffusion of compounds into crystal Often these compounds can reach the active or binding site Often enzymes are active in crystalline state Two types of solvent: ordered and disordered • Ordered water molecules show up as discrete blobs of electron density in contact with the protein or with other ordered water molecules • Disordered water regions show up as featureless (flat) electron density PDB files: • Basically just simple tekst files • At the top: information about the crystal: – Which proteins/ligands etc – Crystalization conditons – How was the structure solved – The resolution – Some usefull statistics to judge the quality of the crystal – How to get from the structure to the biological unit – Remarks about missing bits etc. • Crystal parameters: cell dimensions/space group • A list of all atoms in the structure A crystal structure according to the protein data bank (PDB) occupancy x,y,z coordinates (Å) ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM 25 26 27 28 29 30 31 32 33 34 N CA C O CB CG OD1 OD2 N CA ASP ASP ASP ASP ASP ASP ASP ASP VAL VAL A A A A A A A A A A 928 928 928 928 928 928 928 928 929 929 19.062 19.770 19.075 19.074 21.259 22.112 21.693 23.239 18.417 17.726 9.157 10.123 9.938 8.824 9.776 10.245 11.114 9.742 10.985 10.864 35.067 34.232 32.899 32.351 34.071 35.233 36.025 35.349 32.405 31.125 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 4.73 4.58 4.56 5.39 3.13 5.52 5.42 7.93 3.68 4.63 Isotropic B-factor or temperature factor is a measure of the mobility of an atom B (Å2) = 82<u2>, where <u2> is the mean square atomic displacement N C C O C C O O N C At typical resolutions (1.8 Å or worse) • The electron density of hydrogen atoms is not resolved (and no hydrogen atoms are present in the pdb file) • The electron densities of C, N, and O atoms are all rather similar Position of N and O atoms in Gln (and Asn) side chain must be inferred from hydrogenbonding network Main chain carbonyl Main chain amide ? ? H H H Asp The same holds for the orientation and protonation of the imidazole ring of histidines ? A pdb file may contain residues for which no, or only limited electron density is visible ? ? No density for amide N of glutamine Break in side chain density of glutamate Sometimes the electron density suggests two side chain conformations but often only one is modeled in the pdb file Threonine side chain conformation present in pdb file Alternative conformation that is also compatible with electron density The interpretation of dynamic loops in the pdb file may be tentative Well defined -strand in the core of a protein: atomic positions are reliable Flexible loop at the surface of a protein: atomic positions are not well defined Look at B-factor distribution! Protein coloured by B-factor: Well defined regions have low B-factors (blue/green) Poorly defined/more mobile regions have high B-factors ( yellow/orange/red) A protein molecule is dynamic • The electron density is a spatial average over all molecules in the crystal and a time average over the duration of the X-ray data measurement • Multiple discrete conformations of a residue in different molecules are superimposed. • Damage caused by X-rays may change the protein (mainly breaking of disulfide bonds) • A crude description of dynamics is provided in the pdb file as the isotropic B-factor • Some dynamical aspects evident in the electron density are lost in the pdb file Reading a crystallography paper: Judge the quality of the data: Rmerge: 0.05-0.10 good, 0.1-0.15 acceptable I/σ = signal/noise >2.0 Completeness Redundancy Rwork/Rfree: difference < 0.05, Rwork≈ resolution/10 Deviations of known geometry waters: at 2 Å ≈ 1 water/residue, at > 3Å usually none More and more structures: learn how to use them! Crystallography and drug design and lead optimization The crystal structure of a protein-substrate complex can serve as starting point for structure-based drug design Guanidino group provides additional interactions Relenza was developed starting from the crystal structure of influenza virus neuraminidase with bound sialic acid Structures of bird-flu neuraminidase reveal new cavity that could be exploited in drug design Russell et al., Nature 443, 45-49 (2006) Can X-ray crystallography contribute to lead discovery? - - Development of high through-put (HTP) methods in crystallography has considerably reduced the time needed to solve a crystal structure while minimizing the need for human intervention This now allows for screening of medium sized compound libraries (~1000 compounds) • Library used in traditional HTP screen – 106 compounds – Mw 300-500 Da • Library used in X-ray based screen – 103 compounds – Mw 100-250 Da Technical advances enabling HTP crystallography Automated set-up of crystallization in 96-well format (100 + 100 nl drops Automated imaging of 96well crystallization plates Automated crystal transfer from liquid nitrogen to X-ray beam Stronger in house X-ray sources Automated beam lines at synchrotrons Improved software for automated interpretation of ligand density Expanding hits into larger and higher affinity compounds -Joining fragments by an appropriate scaffold (C) -Grow a fragment to fill neighboring pockets (d) Electron densities of initial fragment (left) and expanded fragment (right) Simultaneous binding of two compounds from a mixture can be detected Top: Electron density of two compounds bound simultaneously together with the two automatically built compounds. Bottom: Electron densities from individual soaks shown in different colors Drug design cycle Crystallise complex Test inhibitor Design better compound in silico Synthesize inhibitors Contribution of X-ray crystallography to drug design and discovery • Lead optimization – Well established – All major pharmaceutical companies do it – Numerous drugs on the market and in pipeline • Lead discovery – Promising, but quite recent – Performed in small companies (that have collaboration agreements with large pharmaceutical companies – Must still prove its value