Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
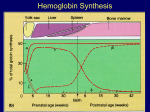
LEARNING OBJECTIVES HEMOGLOBINOPATHIES AND THALASSEMIA Module Objectives: At the end of this module you should be able to Explain hemoglobin and its types. What are hemoglobinopathies? Explain the pathophysiology that causes hemoglobinopathies and thalassemia. Explain how thalassemias are categorized. Describe the basic and specific investigations to rule out hemoglobinopathies and its types. Session Objectives : Correlate the results of laboratory testing with specific thalassemias and hemoglobinopathies. LECTURE OUTLINE HEMOGLOBINOPATHIES AND THALASSEMIA Hemoglobin: One role of blood is to take oxygen from the air in the lungs and deliver it to all parts of the body. Hemoglobin is the part of the red blood cell that carries the oxygen. The most common form of hemoglobin is made up of two parts, alpha globin and beta globin. ¼ million people are born worldwide each year with one of the disorders of the Structure or synthesis of hemoglobins (Hb) Hemoglobin Structure: Normal adult hemoglobin molecule (HbA) consists of two different types of polypeptide chain. Chains are designated alpha and beta. The four chains are folded and fitted to form a globular tetramer with a MW of ~64 500. The structure of Hb A is α2β2. Fetal Hemoglobin predominant hemoglobin in the intrauterine life. At birth it is 80%, starts decreasing after birth, replaced with HbA and Hb A2. In adults : 97% is Hb A & 03% is Hb A2. The composition of Hb F is α2γ2. The composition of Hb A2 is α2δ2. HEMOGLOBINOPATHIES DEFINITION : Hemoglobinopathies are the disorders of globin chain synthesis Classification of Hemoglobinopathies Two main types: i. Qualitative Disorders. ii. Quantitative Disorders. Qualitative Disorders (Structural Variants/ Disorders): Abnormal hemoglobins are formed when the sequence of globin chain amino acids is altered. There is positional substitution of one or more normal amino acids in any of the globin chains. E.g. sickle cell anemia, (valine substituted in place of glutamic acid at the sixth position of beta chain.). It may also result from deletion/frame shift in one of the globin chains. Quantitative Disorders (Expression/Synthesis Disorders): Thalassemias result from a lack of production of particular globin chains to maintain adequate Hb levels. Hereditary persistence of fetal hemoglobin (HPFH) results from a failure to switch from fetal to adult Hb. Hemoglobinopathy Genetics: Homozygous: Inheritance of two genes from each parent coding for the same type of abnormal hemoglobin, e.g., Hb SS Heterozygous: Inheritance of genes from each parent which code for a different type of abnormal hemoglobin each, e.g., Hb SC. THALASSEMIA: Hereditary disorder that can result in moderate to severe anemia. Basic defect is reduced production of selected globin chains. An imbalance of globin chain production results in the accumulation of free globin chains in the red blood cell precursors, which, being insoluble, precipitate, resulting in hemolysis of the red blood cells Underproduction of the alpha globin chain results in alphathalassemia. Underproduction of the beta globin chain results in betathalassemia. Pathogenesis: Genetically determined. Heterogenous group of disorder. Reduced synthesis of one or more types of normal hemoglobin polypeptide chain. Reduced hemoglobin involving affected chain. Normal Hemoglobin: HbA - α2β2 HbA2 - α2δ2 HbF – α2γ2 Each goblin chain has separate genetic control. α –thalassemia affect α-chain synthesis. β –thalassemia affect β -chain synthesis. α-Thalassemia: Definition: An absence or deficiency of α-chain synthesis due to deletion of α-genes. Symbolism: Alpha Thalassemia: Greek letter used to designate globin chain: /: Indicates division between genes inherited from both parents: / • Each chromosome 16 carries 2 genes. Therefore the total complement of genes in an individual is 4. -: Indicates a gene deletion: - / Classification & Terminology Alpha Thalassemia • Normal / • Silent carrier - / • Minor -/- --/ • Hb H disease --/- • Barts hydrops fetalis --/-- Pathogenesis of α-Thalassemia • In normal individual HbA, HbA2 and HbF need α-chain for their formation. • 4 genes of α-chain, each pair on short arm of chromosome 16 present with genotype α, α/α, α. • In α-thalassemia, delation of α-genes reduction or absence of synthesis of α-chain depending on number of α-gene delation. • ↓α-chain synthesis free γ-chain in the fetus & β-chain in infant of 6 months, and continue in the rest of life. • Complementary 4γ and 4β are aggregated (4β), respectively. Hb Bart (4γ) and HbH • Variants of α-Thalassemia • Silent carrier – Deletion of single α-gene – Genotype α/αα – Asymptomatic – Absence of RBC abnormality • Thalassemia trait • • – Deletion of 2 α-genes – Genotype --/αα – Asymptomatic, minimal or no anemia. – Minimal RBC abnormalities. Hb H disease – Delation of 3 α-genes – Genotype --/- α – 75% reduction of α-chain – 25% α-chain synthesis >> small amount of HbF, HbA, & HbA2 – Fetus can survive. – Severe anemia. – Severe RBC abnormalities. Hydrops fetalis – Delation of all α-genes – Genotype --/-- – Absence of α-chain synthesis – Only Hb Bart (γ4) is produced (High affinity for O2 and can not dissociate O2 to tissue). β-Thalassemia Definition: An absence or deficiency of β-chain synthesis of adult HbA. β Chain synthesis ↓ Hb-A ↓ γ and δ chain Hb-A = α2β2 Symbolism Beta Thalassemia • Greek letter used to designate globin chain: +: Indicates diminished, but some production of globin chain by gene: + 0: Indicates no production of globin chain by gene: 0 Classification & Terminology Beta Thalassemia • • Normal Minor • • Intermedia Major / /0 /+ 0/+ 0/0 +/+ • β-thalassemia major – Mutation of normal β-gene β0-gene absence HbA increased HA2 and HbF – genotype – β0β0 – • β-thalassemia intermedia – ↑HbA2 – ↑HbF – ↓HbA – Genotype β+ β+ or β0 β • β-thalassaemia minor – ↑HbA2 – HbA normal – HbF normal Pathophysiology of β-Thalassemia: Various mutations in β-gene ↓ Complete or partial absence of β-chain ↓ Decreased adult HbA ↓ α-chain synthesis remain normal ↓ Free complementary α-chain – unstable and precipitate within normoblasts as insoluble inclusions ↓ Cell membrane damage & impaired DNA synthesis apoptosis i.e. ineffective erythropoeisis 70-80% marrow normoblasts undergo apoptosis Inclusion bearing red cells undergo sequestration & destruction in spleen Special Cases Thalassemia: Hb Lepore: fusion seen in some types of thalassemia Hb Constant Spring: • • chain with 31 additional amino acids --/cs Hereditary persistence of fetal hemoglobin (HPFH) Hb H • • • 4 tetramer Associated with --/- thalassemia Hb Barts & hydrops fetalis • • • • Barts is a 4 tetramer Associated with --/-Lethal High concentrations are capable of sickling LAB INVESTIGATIONS Primary Laboratory Investigation Variable hemogram results proportional to the severity of the thalassemia. • Severe cases present with • • Microcytosis • Hypochromia • Poikilocytosis • RBC counts higher than expected for the level of anemia Findings in severe cases can mimic those seen in other microcytic/hypochromic anemia. • Results of the reticulocyte count are variable • NRBCs may be present (contrast with iron deficiency anemia) Secondary Laboratory Investigation Hemoglobin electrophoresis • Major test for identifying thalassemia and hemoglobinopathy. Types • Cellulose acetate: Alkaline pH • Citrate agar: Acid ph Conclusion: • In retrospect, it is fortunate that the hemoglobinopathies were the first group of genetic diseases to be examined at a molecular level, because they exhibit such a diversity of molecular pathology.