Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
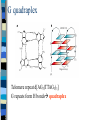
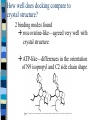
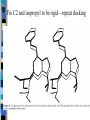
Docking and Cancer Drug Design Jennie Bever Where to start? Ligand (analog-based) Target (structure-based) Goals of Docking 1) 2) 3) Characterize binding site - make an image of binding site with interaction points Orient ligand into binding site - grid search - descriptor mapping - energy search Evaluate strength of the interaction DG bind= DG complex – (DGligand + DGtarget) Structure-based design of selective and potent G quadraplex-mediated telomerase inhibitors Neidle et al. 2001 PNAS 98(9) 4844-4849 NMR-derived G4 structure PDB entry 143D Repeat of d[AG3(TTAG3)3] Telomerase Activated in 80-90% human tumors Keeps cancer cells’ telomeres stable length Telomerase as a Drug Target Telomerase needs single stranded 3’DNA primer end – To hybridize with telomerase RNA template Drug Target: – Use higher order structure to prevent telomeres from unfolding No ss primer Block telomerase G quadraplex Telomere repeat d[AG3(TTAG3)3] G repeats form H bonds quadraplex Stabilize Quadraplex– How? Extended planar aromatic chromophore Nearly all known quadraplex ligands share this structure Problems with known ligands Affinity for duplex DNA comparable to quadraplex binding Low potency for telomerase inhibition IC50=2-5mM Level of acute cytotoxicity too high From Structure to Ligand 1) Energy minimize structures of target and ligand Molecular dynamics simulation time averaged structures 3) Create pseudointercalation binding site Docking Dock ligand into pseudo-intercalation site – Manual, automatic, and flexible ligand docking Energy minimize to determine DG complex Determine DGligand _=interaction energy of ligand with surroundings when explicitly solvated Visual inspection of compound 1 docking Compounds 3 and 4 Similarly docked Docking results Attempts at docking compound 2 were unsuccessful – Distorted the quadraplex structure due to bulky side chains Ligand/quadraplex interactions in solution: SPR Rate constant determination Ligand/quadraplex interactions in solution: SPR Equilibrium constant determination Will these molecules work? Trap Assays and Growth Inhibition (Cytotoxicity) Docking-based Development of Purine-like Inhibitors of Cyclin-Dependent Kinase-2 Koca et al. J. Med. Chem. 2000, 43, 2506-2513 Cdk2 structure complexed with ATP Cdk2 regulates G1/S transition Cdk deregulation in many tumors and tumor cell lines Xray structures of 3 cdk2 complexes available – ATP, olomoucine, roscovitine All bind in same cleft, but with different orientations Docking of 2,6,9-purine derivatives into cdk2 Minimize structures (target and ligands) Represent active site as spheres Calculate partial atomic charges from force fields Rigid docking Pick receptor structure to use Rigid and Flexible docking with proposed ligands Interaction energy and Inhibitory potency (IC50) for 2,6,9-purine derivatives on cdk1 and cdk2 They say good correlation for cdk2 interaction energy can be used to predict activity Interaction energy versus IC50 For cdk1, rank of activities also closely bounded to rank of interaction energies Searching for New Inhibitors Role of Electrostatic and van der Waals contribitions in binding ligand to active site H bonds predicted by rigid docking agree with experimental data How well does docking compare to crystal structure? 2 binding modes found roscovatine-like—agreed very well with crystal structure ATP-like—differences in the orientation of N9 isopropyl and C2 side chain shape Fix C2 and isopropyl to be rigid—repeat docking Paper Conclusions: Rigid docking useful for activity predictions Relationship between cdk1 activity and cdk2 interaction energy Use these experiments to design new inhibitors of cdk2 and cdk1 My Conclusions Docking is now and will be an integral tool in drug design in the future Play with some of the modeling software—it’s cool!—and helps to understand how simulations are done Thanks to Dr. Bourne!