Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
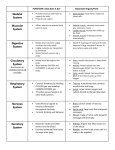
Myotonic Dystrophy Type I (Steinert’s Disease) What is myotonic dystrophy? Myotonic dystrophy type I (DM1), also known as Steinert’s disease, is the most common form of adult-onset muscular dystrophy affecting approximately 1 in 8,000 people worldwide. It is characterized by progressive muscle weakness and difficulty in relaxing the muscles after they have been contracted (myotonia). However, it is misleading to think of myotonic dystrophy as only a muscle disorder because so many of the body’s systems may be affected. DM1 commonly impacts a person’s heart, breathing, vision, gastrointestinal and reproductive systems, as well as cognition. Who can be affected by myotonic dystrophy? Myotonic dystrophy can affect people at any age. The majority of people are diagnosed by the time they reach their early twenties. With each generation, symptoms are more severe than the prior generation. This is known as anticipation. What are the different forms of myotonic dystrophy? Myotonic dystrophy type I (DM1) has two forms: an adult form and a congenital form. Congenital myotonic dystrophy, the most severe form of myotonic dystrophy, is present at birth. It is almost always passed to the child from an affected mother. When the father has myotonic dystrophy, his children are not at risk for developing the congenital form. Babies with congenital myotonic dystrophy are very weak and frequently have problems sucking, feeding, and breathing. If they survive the newborn stage, they generally overcome their breathing and feeding problems, but they are slow to develop language and motor skills and are often affected for life with difficulties in these areas. With the adult form of DM1, symptoms generally appear between the ages of 10 and 40 years. The severity of symptoms, rate of progression, and degree of disability vary widely from one person to the next, even among family members. In most cases, the disorder progresses slowly. Myotonic dystrophy type II (DM2) - also known as proximal myotonic myopathy - is a milder and more rare form of myotonic dystrophy. The disease development and progression resembles DM1, however, unlike DM1, DM2 does not exhibit anticipation (the tendency of the disease to become more severe with successive generations). What causes myotonic dystrophy? In patients with myotonic dystrophy, there is a problem with a particular gene that causes it to convey faulty instructions. This mistake results in National Office: 2345 Yonge Street, Suite 900, Toronto, Ontario M4P 2E5 T 416.488.0030 1.866.MUSCLE.8 F 416.488.7523 W muscle.ca Myotonic Dystrophy Type I (Steinert’s Disease) / page 2 the symptoms of DM. The two types of myotonic dystrophy are caused by mutations in different genes. Although DM1 and DM2 show similar symptoms, they have fundamentally different origins. Type 1. The genes responsible are found on chromosome 19. Each chromosome consists of a long chain of chemicals that form the units of DNA. These units are called nucleotide bases. The disease is characterized by stretches of DNA (abbreviated CTG) on the DMPK (dystrophiamyotonic protein kinase) gene that are repeated several times. It is sometimes referred to as a trinucleotide repeat disease because of the repetition of these three DNA base pairs. In healthy people, there are between 5 and 37 repeats of the CTG sequence. People with myotonic dystrophy type 1 have expanded repeats which can contain anywhere from 50 to more than 4,000 repeats of the CTG sequence. Type 2. The genes responsible are found on chromosome 3. The repeat sequences contain stretches of DNA in which four chemicals (abbreviated CCTG) on the Znf9 (zinc finger protein 9) gene are repeated. As in DM1, the disease occurs after the number of repeats exceeds a certain threshold. Healthy individuals have fewer than 75 CCTG repeats. People with DM2 can have anywhere between 75 and 11,000 repeats. How is myotonic dystrophy inherited? Myotonic dystrophy is transmitted by an autosomal dominant pattern of inheritance. “Autosomal” refers to any of the 22 chromosomes not associated with determining the sex of the child. “Dominant” means that one copy of the mutated gene is enough to cause the disease. There is no carrier status. Each child born to an affected parent has a 50% chance of inheriting mutated gene and developing the disorder, and a 50% chance of being unaffected. The mutated gene can be passed to male and female children with equal frequency. What are the symptoms of myotonic dystrophy? Myotonic dystrophy is a very complex condition. It involves many of the body’s systems, and there are a wide variability of symptoms. Symptoms may include: • Trouble relaxing a muscle (myotonia) • Muscle weakness (myopathy) • Muscle stiffness • Drooping eyelids (ptosis) • Delayed and/or impaired speech • Difficulty raising the head when lying • Difficulty holding or lifting an object • A shuffling gait • Difficulty climbing stairs or rising from a seated position • Cataracts, which develop fairly slowly but can occur in people as young as 30 years • Abnormal heartbeat or dizzy spells • Difficulty swallowing • Bowel problems, such as constipation and stomach pain • Reproductive challenges • Respiratory problems, such as infections and shortness of breath • Premature balding or thinning of hair • Excessive daytime sleepiness How is myotonic dystrophy diagnosed? Diagnosis is made by a physician based on family history, a physical examination and tests, such as: • genetic testing • electromyography (EMG) to measure electrical activity in the muscle • muscle biopsy National Office: 2345 Yonge Street, Suite 900, Toronto, Ontario M4P 2E5 T 416.488.0030 1.866.MUSCLE.8 F 416.488.7523 W muscle.ca Myotonic Dystrophy Type I (Steinert’s Disease) / page 3 Is there any cure or treatment for myotonic dystrophy? Efforts to find a cure for myotonic dystrophy are ongoing. At present there is no treatment that will slow or stop the disease progression. The treatments that are currently available are symptom oriented. For example: • Surgery for correction of cataracts • Medication may be prescribed to counter the effects of myotonia • Medication and/or surgery to address heart problems or excessive daytime sleepiness • Speech and physical therapy • Occupational therapy to assist with daily activities • Mechanical ventilation to support breathing Please visit the myotonic dystrophy section of our website - www.muscle.ca - for more detailed information related to: - Anesthetic Management - Respiratory Care and Management - Occupational Therapy - Physical Therapy What is the prevalence of myotonic dystrophy type I? DM1 is the most common form of adult-onset muscular dystrophy. On average, occurs in 1 per 8,000 live births, but the prevalence is much higher in the Saguenay-Saint Laurent area of Quebec (approximately 1:500). What about research? Current efforts in myotonic dystrophy research are geared to both treatment of the disease and alleviation of symptoms. A number of molecules are under investigation for the relief of muscle spasticity, muscle insulin resistance, and improving muscle function. Scientists searching for a cure are investigating means to eliminate toxic RNAs that accumulate in the cells, or preventing the mutated gene products from binding and impairing the function of regulator of alternative splicing, such as MBNL. Muscular Dystrophy Canada funds myotonic dystrophy research. Please visit www.muscle.ca to learn more about our current research projects. Where can I get more information? For up-to-date research information, please visit: • Muscular Dystrophy Canada: www.muscle.ca • National Institutes of Health: www.nih.gov • Orphanet: www.orpha.net • PubMed: www.pubmed.gov We’re here to help. Contact us at: 1-866-687-2538 [email protected] Join us on social media www.facebook.com/muscle.ca twitter.com/md_canada Disclaimer: This document is intended for general information and awareness. Muscular Dystrophy Canada will not be held responsible for misuse of information or any damages incurred as a result of its use. This resource is not meant to replace consultations with your doctor or to provide medical advice, diagnosis, or treatment. For information specific to the condition affecting you or your family, please consult your physician. National Office: 2345 Yonge Street, Suite 900, Toronto, Ontario M4P 2E5 T 416.488.0030 1.866.MUSCLE.8 F 416.488.7523 W muscle.ca Revised Nov 2013