Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Compounding wikipedia , lookup
Discovery and development of integrase inhibitors wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
Metalloprotease inhibitor wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Drug interaction wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
List of off-label promotion pharmaceutical settlements wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
Drug design wikipedia , lookup
Drug discovery wikipedia , lookup
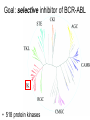
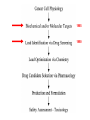
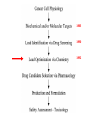
Imatinib pre-clinical and clinical development Stephen Oh, M.D, Ph.D. Markey Program October 30, 2014 Overview • Historical narrative of imatinib development • Paper discussion • Imatinib as a paradigm for other targeted therapies (e.g. JAK2 inhibitors) Inhibiting the kinase activity of BCR/ABL “won’t work” because: • ATP binding pocket of ABL is well conserved among many TKs • Besides, inhibition of BCR/ABL will also inhibit cABL, giving unknown toxicity • What we need is drug to block cancer-specific pathways! Goal: selective inhibitor of BCR-ABL • 518 protein kinases Designing a BCR-ABL inhibitor • • • • • • • How do you screen or design a drug? What preclinical tests do you want? What animal studies do you want? Who are the first patients to try drug? What are endpoints? What to compare to? What is ultimate goal? ~$800 million Discovery starts Year 8 (start Clinical) Year 15 1. Cancer cell physiology • 1960 = Nowell & Hungerford “Philadelphia” chromosome observed, short chr. 22 • 1973 = banding technique enables Rowley to identify Ph chr. = t(9;22) • 1982 = ABL involved on chr. 9, 1984 = BCR gene on chr. 22 • 1990 mouse models validate BCR-ABL is necessary and sufficient for CML development 1985 1990 2. Molecular target: Kinases • 1980 Ciba-Giegy (Novartis) shut down cancer research • 1983 re-opened under Alex Matter • Prior work on interferon convinced him that nature could produce compounds to kill cancer. • Interest in pursuing kinase inhibitors solidified in 1985 Staurosporin inhibits PKC. PKC activity Kinases at Ciba-Giegy (Novartis) • 1985 hired Nick Lydon to head kinase program under Matter. • 1988 Staurosporin derivatives against PKC, but in search of a disease to target kinases. • PDGF-R identified as potential kinase with cancer and cardiology uses, so began search for inhibitor. • Lydon has connection with kinase group at DFCI in Boston. Kinases at DFCI • Lydon meets Brian Druker in 1988 on visit to DFCI where he is post-doc fellow. • Druker convinced BCR-ABL could be targeted after seeing inhibition of EGFR results in Science 1988. • Approaches Lydon about BCR-ABL but CG is hesitant to pursue because small number of CML patients, agrees to include ABL in kinase screening panel. • 1990-1993 DFCI sever ties with CG in favor of Sandoz for kinase work. • Druker has no contact with Matter, Lydon. 3. Lead identification/drug screening • 2 main Questions after identifying targets: – 1. What compounds to test in system? – 2. What is your screening system? • Empiric = NCI uses 60 cancer cell lines. Screens 10,000 chemicals/yr from library in proliferation assay, 500 drugs pass and 5 novel agents recently identified. • Rational synthesis = CG/Novartis approach. 3. Lead identification/drug screening • 1990 chemist Jurg Zimmermann and biologist Elizabeth Buchdunger at CG. • Goal: Rational synthesis to design drug that binds ATP pocket in kinase domain (PDGF-R main target). 3. Lead identification/drug screening • Rational synthesis = Staurosporin derivatives: 1988 inhibit PKC (s), 1990 EGF-R (s), Abl (non-selective) • Screen compounds using in vitro kinase phosphorylation assay against PKC, PKA, EGF-R, PDGF-R, Alb, Src, Lyn, Fgr. • Follow-up with in vitro antiproliferative assay using kinase-transformed cell lines. • 1990 Zimmermann use PAP derivatives to screen for PKC inhibition. 1992 PDGF-R (ns- gets Abl also) = LEAD COMPOUND ID! Phenylamino-pyrimidine (PAP) • Until PAP used hundreds of compounds screened but: lack Abl selectivity, poor “drug likeness” • PAP structure has good Drug likeness: absorb oral, nontoxic, not destroyed in liver, stable in stomach, not excreted too fast. Staurosporin PAP 1985 1990 1992 4. Lead (CGP57148) Optimization • August 26, 1992 first batch of drug. • Buchdunger using in vitro kinase assay in early 1993 inhibits Abl, and PDGF-R. • Spring 1993 CG started to contact physicians for CML interest – NO INTEREST. 1985 1990 1992 1994-5 1994-5 5. Drug candidate selection/production • August 1993 Druker leaving DFCI for OHSU (no longer committed to Sandoz) contacts Lydon at CG for update on inhibitors. • Druker is convinced Abl inhibitors will work. Gets 4 drugs from CG to test on BCR-ABL using protein, cell and animal experiments. • Feb. 1994 presents results to CG = 90% inhibition of BCR-ABL in vitro and picks CGP57148 as best drug to pursue for CML. Nature Medicine, 1996 Fig. 4 In vivo antitumor activity of CGP 57148 Fig. 5 Colony-forming assays 1985 Typical 8yr 1990 1992 1994-95 1994-95 1995-97 Animal safety/toxicology • 1995 rodent studies no problems with IP delivery. • March 1996 Ciba-Geigy and Sandoz merge to become Novartis and new management take over. • 1996 dog study problem. Clots develop at IV catheter site entrance. • Phase I planning slowed at Novartis. • Nov. 1996 rats develop liver toxicity and all human/animal trial planning stopped. • 1997 Druker convinces Novartis to continue with STI571 and drug is made orally bioavailable. • Rats and dogs absorb oral formulation. • Monkeys only got liver toxicity at “hi” doses. • Decided to proceed to human studies with same formulation, despite rat/dog toxicity. 1985 1990 1992 1994-95 1994-95 Year 8 (start Clinical) 1995-97 1998-2000 (Imatinib = 3yrs.) 1999-2001 2000 Year 15 Apply 2/27/01approved 5/10/01 Cancer clinical trials - Phase I • Phase I = What is the tolerable dose of new drug for phase II studies? • Typically not tumor specific, 10-30 patients • Patients with advanced disease, resistant to standard therapy, and good organ function • Dose escalation, looking for acute toxicity. • 3-6 patients at each dose • DLT = 33%, Rx. 3 more patients at same dose. STOP if toxicity, go up if not • DLT > 33%, STOP • Use highest dose with DLT < 33% for phase II At 300 mg or higher, 53/54 (98%) with CHR STI571 – Phase I • Novartis now has to mass produce for anticipated demand – this has never happened for a drug so early. • Plans to scale production from 50 kg in 9/99 to 23 tons in 2001. Cancer clinical trials - Phase II/III • Phase II = Does drug have activity against specific tumor? • Tumor specific study. • Pick patients that are active (good performance status) and minimal prior chemotherapy. • Phase III = Compare efficacy of new drug to standard of care in order to help physicians make treatment decisions. • Randomized, broad eligibility better, multiinstitutional – applicable to “community” doctors. • Endpoints usually survival or symptom control. • 532 chronic phase IFN failures • 400 mg imatinib daily • Complete hematologic response: 95% • Major cytogenetic response: 60% • Median 18 month f/u, 89% still in chronic phase, 95% alive • 2% d/c due to adverse events, no treatment-related deaths FDA approval May 2001 based on Phase II data • 1106 patients randomized • Complete hematologic response: 95% vs 56% • Major cytogenetic response: 85% vs 22% • Complete cytogenetic response: 74% vs 9% Drug discovery cost analysis 6 yrs DiMasi et al, J Health Econ. 2003 Mar;22(2):151-85. Discovery to phase I = 4.3 years 12 year process Phase I to FDA approval = 7.5 years Imatinib = 10 years Drug discovery cost analysis DiMasi et al, J Health Econ. 2003 Mar;22(2):151-85. Methods in this analysis have been criticized – other estimates range widely: $55 million to $2 billion! Imatinib costs • Interferon about $1,700-3,300/month • Initial cost ~ $2,200/mo INCOME COST < 43,000$/yr free 43-100,000$/yr 20% of income >100,000$/yr Full price • Price has more than tripled since initial approval ~$100k/yr • Revenue for imatinib in 2012 ~$4.7 billion Generic Gleevec • Initial patent application filed in 1993 – Did not claim any specific salts or mention imatinib mesylate • Patent application filed in 1998 specifically mentioned beta crystalline form of imatinib mesylate • After lengthy delay, application in India rejected in 2006 – ruling that imatinib mesylate was already known prior to development of Gleevec • Appealed to Indian supreme court – rejected April 2013 • Gleevec to go off patent in US July 2015 • Generic Gleevec to become available in US Feb 2016 Generic Gleevec Imatinib as a paradigm for targeted therapies in hematologic malignancies BCR-ABL negative MPNs Activation of JAK-STAT signaling in MPNs JAK2 V617F JAK inhibitors approved/in development for MPNs JAK inhibitor JAK2 IC50 JAK selectivity Non-JAK targets Clinical trials Ruxolitinib FDA approved 4.5 nM JAK1 0.6x JAK3 72x TYK2 4x Fedratinib Phase III completed 3 nM JAK1 35x JAK3 332x TYK2 135x FLT3 RET MF PV ET Momelotinib Phase III ongoing 18 nM JAK1 0.6x JAK3 8.6x TYK2 Unk JNK1 CDK2 MF Pacritinib Phase III 22 nM JAK1 58x JAK3 24x TYK2 Unk FLT3 MF CEP-701 Phase II 1 nM JAK1 Unk JAK3 3x TYK2 Unk FLT3 TrkA MF PV ET LY2784544 Phase I/II ~60 nM (JAK2 V617Fselective) JAK1 Unk JAK3 15x TYK2 Unk Unknown MF PV ET MF PV ET JAK inhibitors approved/in development for MPNs JAK inhibitor JAK2 IC50 JAK selectivity Non-JAK targets Clinical trials Ruxolitinib FDA approved 4.5 nM JAK1 0.6x JAK3 72x TYK2 4x Fedratinib Phase III completed 3 nM JAK1 35x JAK3 332x TYK2 135x FLT3 RET MF PV ET Momelotinib Phase III ongoing 18 nM JAK1 0.6x JAK3 8.6x TYK2 Unk JNK1 CDK2 MF Pacritinib Phase III 22 nM JAK1 58x JAK3 24x TYK2 Unk FLT3 MF CEP-701 Phase II 1 nM JAK1 Unk JAK3 3x TYK2 Unk FLT3 TrkA MF PV ET LY2784544 Phase I/II ~60 nM (JAK2 V617Fselective) JAK1 Unk JAK3 15x TYK2 Unk Unknown MF PV ET MF PV ET COMFORT-I COMFORT-I Gleevec redux? Not so fast… • No improvement in anemia • No improvement in marrow fibrosis • Modest (at best) improvement in JAK2 V617F allele burden Can these inhibitors selectively target and eradicate the “malignant clone”?