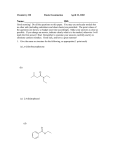
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Microdose: The New Drug Development Approach Tania Perestrelo Msc Toxicology & environmental health University of Utrecht Supervisor: Wouter Vaes & Babs Fabriek (TNO) 2nd Reviewer: Bas Blauwboer (IRAS) 1 Table of content Summary pg 3 Introduction pg 4 Traditional drug development process The microdosing concept AMS PET Predictability of pharmacokinetic pg 8 pg 11 CREAM EUMAPP PBPK modeling technique Pediatric drug development pg 19 Traditional dosage correction Differences in children vs adults pg 21 Absorption Distribution Metabolism Elimination Dosage & effects of drugs in children pg 27 Paracetamol Conclusion pg 28 Discussion pg 29 Reference list pg 31 2 Summary The development process of new drugs is very complex, expensive and time consuming process. The development process consists of several steps, including animal studies and human clinical trials. The process relies in general on the understanding of the metabolism pathways and the pharmacokinetics of a drug. During the development process approximately 40% of drug candidates that enters the clinical trials fail to pass through the first human studies (phase 1). Reason for most of the failures is due to inappropriate toxicological and/or PK properties, therefore predetermining the PK value in humans before the drug is tested in the first human clinical trials will lead to a reduced amount of drug failures. Minimizing the amount of failures will also result in a reduction of the drugs developmental cost, from which approximately 75% was based on drug failures in the early stages of the process. Microdosing is a new promising approach that may help reduce both the amount of animal studies needed and the drug development costs. The approach consists of determining pharmacokinetic and pharmacodynamic properties of the candidate drugs in humans in the early phase of the developmental process. In the study a very small dose of the drug defined as less than 100 µg will be administered to human subjects, following the collection of samples in different time intervals and/or the real-time scan of the patients to observe how the compound is distributed in the body. Drug candidates with better PK properties will be selected for further study, whereas the other candidates will be banned. For the practice of microdose study there are ultrasensitive analytical methods needed. The accelerator mass spectrometry (AMS) and the positron emission tomography (PET) are needed to determine the pharmacokinetics and pharmacodynamics of the candidate drugs in humans. Projects such as the CREAM and EUMAPP confirm the predictability of the microdose to determine PK values related to the therapeutic dose. Pediatric drug development process is very complicated, because children are less likely to participate in clinical trials due to ethical issues. Traditionally the dosage of drugs administered to children is obtained from correction of the adult therapeutic dose with bodyweight and body surface area. This indicates that there is the possibility that children could be underdosed or overdosed in relation with the related effect exerted by the drug. A proper dosage of drugs could be obtained by applying the microdose study in children. 3 Introduction The introduction of new drugs to the market compromises a very long, complex and expensive development process 1. The baseline for the development process of drugs consists of understanding the mode of action, toxicological effect, metabolism and pharmacokinetics of a drug. Traditionally, all data needed to determine among others the toxic effect is obtained from experiments performed on in-vivo, in-vitro and in-silico models. For example the toxicological data such as the disposition of the drug and also the effect of the drug at target organs is obtained from a well-conducted study, which is then analyzed properly before the drug can further be tested on humans in a clinical trial. Understanding the pharmacokinetics (PK) and pharmacodynamics (PD) of a drug is also of great importance for the development process of drugs, for it can predict the possibility of the drug to succeed in performing the desired mode of action. The PK and PD properties determine the absorption, distribution, metabolism and excretion (ADME) characteristics of a compound 1. The amount of drug available to perform a mode of action can be determined or evaluated from the ADME characteristics of a drug. Therefore drugs with better PK and PD properties are more likely to succeed in the development process and will eventually be further tested in the clinical trials, whereas for the others drugs the development process will end. Besides the PK and PD properties also toxicological information such as the no-observed adverse level (NOAEL), effect dose (EC), toxic dose (TD) and lethal dose (LD) is obtained from the performed experiments/ These data are then used to determine the therapeutic index and margin of safety of the drug. The therapeutic index (TI) is determined by the ratio of the required dose to produce a toxic effect and the dose needed to obtain the desired therapeutic response. The most commonly used dose for the index of effects is the median dose, independent of its beneficial or toxic effects. The median dose is referred as the dose required to induce an effect in 50% of the population or the dose that induce 50% of the maximum response. Therefore the TI is calculated by the following equation TI= LD50 / ED 50. The median dose however does not consider the slopes of the doseresponse curve for the therapeutic and toxic effects, which indicate how fast a response increases as result of increased dose. This questions the safety aspect of the drug in an increased dosage. A way of resolving the problem is by determining the margin of safety for the drug. The margin of safety is then calculated by dividing the dose of the drug needed to obtain an undesired effect (LD1) by the dose needed for the desired effect (ED99). Hereby LD1 is referred to the dose that produces a lethal toxic effect in 1% of the population and ED99 is referred to the dose required to induce an effect in 99% of the population 4. The different data obtained from the experimental settings is then extrapolated for prediction of the drug in the human population. Extrapolation consists of dividing the data by uncertainty factors, which takes several variations into account such as the intra- and interspecies variation. In other words the potential differences between the species and within the (human) species itself is taken into consideration. Extrapolation occurs by the mathematical modeling process called allometric scaling, with whom the values for the human population are predicted. After the extrapolation step the candidate drugs are tested in a clinical setting on humans. The clinical trials can be divided in 3 phases: 1) phase I, 2) phase II and 3) phase III. In the human clinical phase I researchers will test the drug on a small group of people (2080) to evaluate the safety of using the drug and also to determine a safe dosage range. In this development phase also the possible side effects of the drug can be observed. If the drug is considered safe it is further tested in the phase II clinical trial to evaluate its safety and also determine its effectiveness in a larger group of people (100-300). After passing these trials the drug is then tested on a larger group of people (1000-3000) in order to confirm the effectiveness of the drug. In these phase the drug effect will be also compared to the effects observed by other commonly used treatments. The possible side effects of the drug will also be monitored. In general all data regarding the safety use of the drug will be collected. After passing the third human clinical phase study the drug will be first submitted for registration and after approval will be introduced in the market and become 4 available for the patients. Hereafter the drug can still be post-markedly tested in studies to give additional information regarding its risk, benefit and optimal use. Approximately 40% of drug candidates that enters the clinical trials during the development process fail to pass through the first human studies (phase 1) 5. The reason for most of these failures is due to toxicity problems and most likely inappropriate PK and metabolism properties. It is therefore of great concern to estimate whether the metabolism pathway and PK of a drug can differ between those in humans and the ones predicted in the model studies, despite the well established extrapolation process which includes the correction for several uncertainty factor. This raises the question: How well can the selected model predict the human situation? Reports can be found in the literature database illustrating how the bioavailability of various drugs can differ between species (Table 1). This emphasizes the difficulty of selecting the appropriate candidate that should be used as model for human predictions 1, 2. The study compared the data of 25 newly developed drug candidates obtained from animal studies to establish the differences in oral bioavailability between species. These drug candidates were ready to enter the human phase 1 clinical trial. The bioavailability of the compounds resulted to be highest in dogs for 32% of compounds, whereas 16% of the compounds had high bioavailability in rats and none in monkeys. This indicates that there is probably no better model for the prediction of pharmacokinetics of a drug in the human population than the human itself. Figure 1 also illustrates that there is no correlation between the bioavailability in humans and animals. The lack of correlation is most probably due to differences in physiology, which can be described by differences in absorption and metabolism, etc 6. Therefore as already mentioned an accurate prediction of pharmacokinetics in human is not possible from animal data. 5 Predetermining the PK value in humans before the drug is tested in the first human clinical trials will minimize the amount of drug failures during the development process, which also results in a reduction of the drugs developmental cost. Approximately 75% of the development costs are based on drug failures in the early stages of the process 1. Therefore by reducing the costs more drugs can be tested for the same or lower costs, which suggests that a variety of new medicines can be placed in the market for the patients and maybe also at a lower price. Here for a new experimental approach called microdosing has been introduced aiming to solve the problems of drug failures. The microdosing approach consist of obtaining the information needed to enhance the probability of the drug development success before entering the clinical phase 1 human study (Figure 2) 1 . With this approach the precise value of the drug in humans is determined by obtaining the human PK values in less time than is needed while conducting a phase 1 human study. In other words it can establish whether the development of a drug should continue or not, according to the obtained PK values for the specific drug 1-3. Traditionally potential drug candidates with high affinity towards a target protein are identified in the early stages of the drug development process. The pharmacological activity of the drug candidates is then confirmed by in-vitro and animal studies, which will finally be tested in a clinical trial starting with the human phase 1 study. With the new approach however the candidate drugs are tested in a microdose study (also known as the human phase 0 study) prior to human phase 1 study of the clinical trial. Small doses of the drugs candidates will be administered to the human subjects to determine the pharmacokinetic and pharmacodynamic properties in the human data 1. Data such as clearance, absorption, distribution, metabolism and half-life of a drug can then be obtained in a much earlier stage compared to the traditional drug development process. This indicates that the best drugs can be filtered out for further investigation. Early human pk data can also be used to determine the best candidates or model for the prediction of human values for further research, suggesting that the amount of animal studies needed for a specific research can be reduced. Figure 2: A schematic view of the traditional drug development process and the new microdosing approach 1 . Traditionally the human data of the drug metabolism pathway and pharmacokinetics can only be obtained when the drug is tested in human phase 1 studies for approximately 18 months, whether with the new approach the data can be obtained within 4 months. Hereby it can estimate the precise value of the drug before continuing a very long and expensive development process. 6 In general, microdosing is a promising approach that helps reduce both the amount of animal studies needed and the drug development costs. By lowering the drug development costs it also facilitates the introduction of new life-saving medicines into the market at lower costs. This makes microdosing an interesting approach for several pharmaceutical industries. In order to better understand the microdosing approach, this thesis will briefly summarize the concept together with its advantages and disadvantages. As already mentioned with the aid of microdosing both the pharmacokinetic and pharmacodynamic properties of a drug can be determined. Therefore the data obtained from the microdosing studies can be analyzed to determine if the obtained values are in accordance with the known values for therapeutic dosage in the literature. If these values for the therapeutic dose can be predicted by a microdose, than it could be questioned whether microdosing can be applied for the prediction of PK values in children. The PK values of adults and children are more likely to be different because of the maturity differences of certain processes as for example the metabolism pathway. Doses for children are mostly obtained by adjusting the normal dose of an adult subject for the height and the weight of the young patient. Therefore applying microdose studies in children may help to establish whether the calculated dosage is actually the right dosage for the children after all. This will also be addressed in the thesis. 7 The microdosing concept Microdosing is a new experimental approach that has been recently introduced as the promising most successful method for the development of drugs. As already mentioned in the introduction most drugs fail to pass through human phase 1 study during the drug development process due to among others inappropriate pharmacokinetic (PK) properties. With the aid of microdosing (or human phase 0) study these properties can be obtained much earlier in the developmental process of the drug without having to go through the long and expensive human phase 1 study. The proper value of the drug can then be estimated before starting the whole phase 1 study and thus determining if the drug should be further tested or not. Drugs that are most likely destined to fail due to inappropriate PK properties will then be banned from further research. Microdose as the name already implies is a very small dose, defined by both the European agency for evaluation of medicinal products (EMEA) and also the Food and drug administration agencies (FDA) as 1/100th of the predicted pharmacological dose derived from in-vitro and animal models or 100 micrograms 5,1. From the definition of a microdose from the agencies it can already be noticed that it is still not exactly clear what the real dose should be for a microdose study. The agencies however also stipulates that the smallest dose of the drug candidate, whether the 1/100th of the predicted dose or 100 micrograms should be taken into consideration for the human microdose study 1. This very small dose is considered to be much safer than the pharmacologically active dose and is therefore considered safe to start testing the candidate drugs in a human phase 0 clinical study with such low level of drug concentration. Although the dose is considered safe for testing in a human phase 0 study, it is important to elicit that no efficacy or safety data can be obtained from such study due to the lower dose used. Therefore a conventional phase 1 study should always be conducted to demonstrate the tolerability and safety of subjects to the candidate drug 1. Before any human clinical trials including a microdose study can be carried out the candidate drug should have been tested on several studies in advance ensuring that exposure of human subjects to the drug during the study will cause no harm to the subjects (Table 2) 1. The safety evaluation of nonclinical studies as referred in the M3 guideline of the international conference on harmonization of technical requirements for registration of pharmaceuticals for human use (ICH), should estimate the initial safe starting dose and dose range for human trials and also identify parameters for clinical monitoring of possible adverse effects 25. Therefore as expected one of the studies consists of determining the safety margins, which is established by estimating the no-observed effect level and the maximum tolerated dose in a single species. Drug candidates that pass these tests with proper results are considered safe and can then be further investigated in a human microdose study. Subjects in a microdose study are exposed to very low dose concentration of a compound, indicating that the presence of ultrasensitive analytical methods capable of measuring concentration range of pictogram to femtogram are of extreme importance 1. The only ultrasensitive analytical methods nowadays available that can be used in a microstudy are the accelerator mass spectrometry (AMS) and the positron emission tomography (PET). With the aid of the AMS in a microstudy the pharmacokinetic data of a compound can be determined, whereas the PET can be used to analyze the pharmacodynamics. For the AMS microstudy body fluids are analyzed to determine the concentrations of the drug and metabolites, which is the used to estimate the PK properties of the compound. These fluids are in general as for most analysis collected from the exposed subjects at different time intervals after dosing. For the analysis of pharmacodynamics on the other hand, real-time data is needed. Therefore subjects have to go through a complete PET body scan after exposure, which will illustrate the disposition of the drug through the body 1, 6. Usage of these ultrasensitive analytical methods however are only possible if the drug candidate that is administered to the subject is labeled. The drug can be labeled in two ways: 1) attaching an isotope-ligand or 2) the drug itself can be radiolabeled. The most frequently used technique is labeling of the drug with an isotope-ligand. This is a much easier way of labeling a drug compared to the difficulty of synthesizing a radiolabelled drug itself. 8 Table 2: Proposed toxicology tests to permit a human Phase 0 microdose study AMS microdosing A small group of human volunteers are exposed to a single dose of 14C-labelled drug in a concentration range of 1-100 micrograms either intravenously or orally through ingestion of the drug. The study design is a randomized control trial, in which subjects are exposed in a parallel or cross-over manner to a compound followed by a washout period 1. After exposure samples of blood and urine is collected from the volunteers during a 60-hour period, which should be sufficient to retrieve plasma half-lives of the administered drug. The concentration of the total drug or metabolite is obtained from the blood samples, whereas the amount of drug excreted from the body through urine is obtained from the urine samples. After sampling the compound is extracted from the samples and chromatography separated in order to obtain sample fractions that are analyzed by the AMS to detect and quantify both the parent compound as the metabolite. The advantage of the AMS technique is that from a single run both information about the parent drug and its metabolite can be obtained. With this information the capability of humans to metabolize the compound can be determined, which can be used to rank compounds in an early stage of drug development. Ranking of the compound according to the metabolizing capacity of humans should reveal compounds with promising features depending on the desired effect of the designed drug. For example if the drug was designed to reduce pain by blocking a receptor, than the compound should not be easily or rapidly metabolized for it will only result in minor effect. These way drug candidates with better properties can be selected to continue further in greater clinical trials during the drug development process. PET microdosing Subjects participating in the microdose study will be administered a single dose of radiolabeled drug, followed by a non-invasive scan by the positron emission tomography (PET). This scan as already mentioned illustrates the disposition of the compound in the body, determining therefore the pharmacodynamic properties of the compound. In general, results obtain from the PET-scan informs about the ability of the drug to reach the target tissues 1. Distribution of drugs in the body is of great interest for it determines the target organs that will be affected. Therefore also information about possible adverse effects due to the effect of the drug on non-desired target organs can be obtained. For example the nervous system such as the brain, distribution of a drug in the brain is of great interest for the determination of possible side effects such as sleepiness. In general PET imaging can be used to determine the distribution of the drug in the body, illustrating the amount of drug that reached a specific target. The amount of drug in the image can therefore not be distinguished as bound or unbound drug in the tissue, hence the total effect of the drug on the target cannot be predicted. Therefore the images can only predict whether the drug will reach a specific target organ or not. This is also of great importance for ranking drug candidates with the promising features the drug was designed. For example if the drug was designed to target the brain in the case of psycho- 9 medications it should be shown in the PET images of the head, otherwise it does not reach the target and is therefore destined to fail in the drug development process. As already mentioned above the results obtained from a microdose study can be used to rank drug candidates according to the desired pharmacokinetic and pharmacodynamic features. Ranking of the compounds should facilitate the selection of the drug candidates that are most likely to succeed in inducing the desired effect it was designed for. Selection of candidate drugs before starting a phase 1 clinical study should reduce the costs and time needed for the development of drugs, because only candidates with the proper features will be further tested whereas for the other compounds the developmental journey ends. 10 Predictability of pharmacokinetic The dose in a microdose study is not a therapeutic dose, suggesting already that there might be differences in the estimated PK values from both doses. At therapeutic dose the metabolic enzymes and drug transporters may become saturated which can influence the estimation of the PK value. This is certainly not the case when using a microdose, which raises the concern if a microdose can properly predict the values from a therapeutic dose 2. Therefore it is of great importance to determine if the pharmacokinetics obtained from a microdose can successfully predict those from the therapeutic dose. Several studies attempted to try justifying the microdosing approach, including the well-known studies the European Union Microdose AMS Partnership Programme (EUMAPP) project and the Consortium for Resourcing and Evaluating AMS Microdosing (CREAM) study. EUMAPP A project funded by the European union as an international multi-centre research study that involves the collaboration between industry and academia. Candidate drug selection for the study was based on the known prediction problems the drugs have when using the traditional models (e.g. in-vitro and animal species) to determine the pharmacokinetics 5. 7 drugs were selected, consisting of 6 generic drugs and one failed development drug named S-19812. Table 3 summarizes the selected drugs and their main problems impeding any pharmacokinetic prediction 5. The synthesized drugs were isotopically labeled with 14C and a microdose of 100µg, 7.4 kBq (200nCi) was administered either by oral or through intravenous route (through an infusion over 30min) to the study group, which consisted of 6 healthy male volunteers. Depending of the drug tested a different study design was used differing in the routes of exposure. According to the study design the dose could be 1) orally administered, 2) intravenously administered or 3) administered by a combination of both routes. All drugs were exposed both orally and intravenously in different period of time, with the exception of the drug phenorbital that was only exposed orally. All drugs, with the exception of phenorbital and paracetamol was tested in a 3-way cross-over study, which included besides oral and intravenous exposure also of the combined exposure consisting of an oral exposure with the unlabelled drug and intravenous exposure with the 14C labelled drug in the same period of time. In the combined exposure study design, the intravenous exposure with the labeled drug acts as a tracer. The dosing pattern used for the EUMAPP study is summarized in table 4. Because of the different route of exposure studied it is possible to compare the similarity of the PK data obtained from the microdose (period 1) with that obtained from the therapeutic dose (period 3). Also because of the combined exposure from different routes of exposure, the bioavailability of the drug can also be determined in a single clinical study. Samples were taken from all subjects and also analyzed. CREAM A project with the primary goal of assessing how good a microdose can predict the pharmacokinetics of a therapeutic dose. The candidate drug selection was based on the known problems the drugs have impeding a proper estimation of the PK data. 5 drugs were chosen for the study, consisting of 4 drugs that can found in the market and 1 failed development drug named ZK253 (Table 3) 8. A cross-over study design was chosen for the project. The drugs were isotopically labeled with a radioactive carbon (14C) and were administered orally or intravenously to the study group, which consisted of 6 healthy volunteers for each compound. The dosing regimen used in the project is summarized in table 4. All drugs were exposed both orally and/or intravenously in different period of time, with a wash out period of 14 days between each dose administration time. Besides the oral and intravenous exposure of the compounds ZK253, midazolam and erythromycin, the volunteers have also been exposed to the drugs through both routes of exposure simultaneously. These volunteers received a 14C-labeled intravenous microdose and also an unlabeled oral therapeutic dose at a certain time. Samples were taken from the subjects and further analyzed. 11 Table 3: List of drugs tested in EUMAPP and CREAM study and the possible problems impeding an accurate prediction of the PK data. Study Drug Fexofenadine EUMAPP Paracetamol (acetaminophen) Phenorbarbital Sumatriptan Propafenone Clarithromycin S-19812 CREAM Warfarin Midazolam Erythromycin Diazepam ZK253 Problem impeding PK predictions A probe P-gp and OATP substrate. The Limited absorption is partially controlled by P-gp and it is excreted in urine principally as the parent drug. Primarily metabolized by sulphate and glucuronide conjugation. The metabolites are difficult to quantify from in vitro-data. 100% orally bioavailable, with very low clearance and moderate volume of distribution justifying its long half-life time of approximately 100 hr. Low oral bioavailability (ca 15%) and metabolism-dependent elimination. Clearance and first-pass loss in humans are difficult to predict from in-vitro data. Saturable first-pass metabolism resulting in a dose-dependent PK at therapeutic doses. P-gp substrate undergoes metabolism and exhibit limited oral bioavailability (ca 50%). Trial drug forms an active form S-32361. The ratio of these two are significantly different between data obtained from human phase-1 study and those from animal and in-vitro studies. 100% orally bioavailable with low clearance. Difficulty in determining clearance in human in-vitro systems. Low bioavailability due to extensive first-pass CYP-P450 metabolism. Known substrate for CYP3A. PK in influences by transporters e.g. Pglycoprotein. Low clearance and moderate tissue distribution. Primarily metabolized by CYP enzymes. Oral absorption is uncertain due to conflicting animal data. The pharmacokinetics of a drug obtained from a microdose study can in general be compared to those reported in the literature. Pharmacokinetic data for most of therapeutics used can be found in books. By comparing both data an overall conclusion regarding the reliability of the microdosing approach can be taken. Besides these 2 major projects, there are also several other studies that were carried out to determine the predictability of PK data of therapeutic dose by a microdose for a specific compound. However the same persons Lappin and Garner that are promoting the microdosing approach writes most of the microdosing studies. Therefore only the compounds tested in the EUMAPP and CREAM projects will be further referred to in this paper. The Pharmacokinetic data of the compounds tested in the projects are summarized in table 5, including data of these compounds published in books. The general rule that is applied to predictions obtained by allometric scaling of animal data to human data is also applied to the data obtained from microdose study or literature, which implies that the difference between the data obtained from literature and/or prediction can only vary within factor 2. In other words prediction of the PK from a microdose study is only accepted if the difference between the pk obtained from the study and the literature is less or equal to factor 2. The results of both projects predicted similar pk data as described in the books. Most of the results differed by a factor 2 from the literature data, indicating that there is a good concordance between the pharmacokinetics of a microdose and that of a therapeutic dose. Hereby both projects ensure the predictability of the pk of a pharmacological dose by a microdose, which justifies the approach. This clearly shows that microdosing can be used for the selection of drugs in the earlier phase of drug development. 12 Table 4: Clinical dosing regimen used in EUMAPP and CREAM CREAM EUMAPP Study Drug Clarithromycin Dose period 1 14 C oral (100µg) Paracetamol 14 C oral (100µg) 14 C intravenous (100 µg) Fexofenadine 14 C oral (100µg) 14 C intravenous (100 µg) Phenobarbital 14 C oral (100µg) Sumatriptan 14 C oral (100µg) 14 C intravenous (100 µg) Propafenone 14 C oral (100µg) 14 C intravenous (100 µg) S-19812 14 C oral (100µg) 14 C intravenous (100 µg) Warfarin 14 C oral (100 µg) oral (5 mg) Erythromycin 14 C oral (100 µg) Diazepam 14 oral (2.50 mg) + 14 C intravenous (100 µg) 14 C intravenous (10 mg) Midazolam 14 C oral (100 µg) 14 C intravenous (100 µg) ZK253 14 C oral (100 µg) 14 C intravenous (100 µg) C intravenous (100 µg) Dose period 2 14 C intravenous (100 µg) - Dose period 3 14 C tracer dose + oral therapeutic dose (250mg) C tracer dose + oral therapeutic dose (120mg) 14 C tracer dose + oral therapeutic dose (50mg) 14 C tracer dose + oral therapeutic dose (150mg) 14 C tracer dose + oral therapeutic dose (100mg) 14 oral (7.5 mg) + 14 C intravenous (100 µg) oral (50 mg) + 14 C intravenous (100 µg) 13 Table 5: Pharmacokinetic properties of therapeutic drugs tested in the EUMAPP and CREAM project Drug Study Route Dose Clarithromycin EUMAPP Diazepam Literature 1 Literature 2 CREAM Oral Oral IV Oral Oral IV IV 250 mg 100 µg 100 µg 250 mg 500 mg 100 µg 10 mg Literature 1 Literature 2 Oral Oral 10 mg 2 mg Erythromycin CREAM Fexofenadine Literature 1 Literature 2 EUMAPP IV Oral Oral Oral Oral Oral IV Oral Oral Oral IV 100 µg 250 mg 250 mg 250 mg 120 mg 100 µg 100 µg 60 mg 120 mg 5 mg 5 mg Oral IV Oral Oral Oral IV Oral Oral 7.5 mg 100 µg 10 mg 2 mg 100 µg 100 µg 100 µg 1400 mg 325 mg 100 µg 90 mg 50 mg 150 mg 100 µg 100 µg 50 mg 100 µg 100 µg 200 mg 3 mg 100 mg 25 mg 100 µg 5 mg 6.1 mg 5 mg Midazolam Literature 1 Literature 2 Schwagmeie r (1998) CREAM Paracetamol Literature 1 Literature 2 EUMAPP Tozuka 2010 Literature 1 Propafenone Literature 2 EUMAPP Literature 1 Literature 2 EUMAPP Sumatriptan EUMAPP Warfarin Lacey (1995) Literature 1 Literature 2 CREAM Phenobarbital Literature 1 Literature 2 Oral Oral Oral Oral Oral Oral IV Oral Oral IV Oral IV Oral Oral Oral Oral Oral Oral T1/2 (h) 3.4 4 4.1 3.3 4.5 45. 1 35. 7 43 32. 9 2.5 2.5 1.6 1.4 12 16 8 14 5.1 143 181 .5 3.3 2.6 1.9 1.2 5.8 4.6 2.4 2.0 Cmax (ng/ml) 958 188 1200 4.7 322 V (L/kg) 1.9 2.6 3.3 1.3 1.8 CL (L/h/kg) 0.3 0.4 0.5 0.02 0.02 Fa (%) 39 22 55 50 - 317 - 1.1 2.0 0.02 0.04 100 100 3.4 717 900 318 0.3 286 55.9 - 1.1 0.8 0.6 1.7 4.6 - 0.3 0.2 0.3 0.2 0.6 1.0 5.2 14 35 40 30 41 74.5 - 34 3.0 78 1.1 1.6 0.02 1.1 1.1 3.3 1.8 1.0 0.3 0.4 0.8 0.3 0.3 0.3 22.1 44 88 88 2.5 108 99 90 2.6 3.8 5.4 1.4 1.9 6.5 2.9 1.6 1.0 2.5 274 48 37 46 2.6 98 0.02 26 0.1 95 77 54 5.0 493 500 - 1.1 0.5 0.7 3.9 6.1 2.4 2.0 2.4 1.0 0.3 0.1 0.1 0.3 0.003 0.06 0.7 0.6 0.2 1.0 1.3 0.7 0.002 0.004 0.003 0.002 85 100 90 13 5.8 7.6 20 14 14 15 93 90 a) F defines the absolute bioavailability and is shown in percentage (%) Literature 1 8. Literature 2 9. 14 The microdosing approach is very promising for the selection of candidate drugs with the desirable human ADME properties in the early phase of drug development. Hence only compounds that are capable of inducing a desired response or effect is tested in the human phase 1 clinical trial. With the microdose study the pk properties of a compound can be predicted, however it should be noticed that this predicted value might differ from the therapeutic value because the compound can for example bind to an enzyme or transporter. In this case saturation of the enzyme or transporter plays an important role and will affect the bioavailability of the compound. The bioavailability of a compound is of great importance for it is the free amount of concentration that is available to induce an effect at the target organ or tissue. The doses applied in a microdose study is very low compared to the therapeutic dose, indicating that saturation of enzymes or transporters will not take place and thus in such a case a good indication of the pk paramaters cannot be given. In these cases the data obtained from a microdose study can still be used to predict proper PK values with the aid of other studies such as for example the physiologically based pharmacokinetic (PB-PK) computer model. Figure 3 : Schematic view of PBPK model analysis simulating PK profiles of a drug The physiologically based pharmacokinetic (PBPK) computer model is a mathematical modeling technique, in which all type of dosage forms and delivery methods can be used to predict the time-course behavior of compound in biological matrices (for example blood, urine, exhaled air, tissues) of humans or animal species 6,26. The model is based on a multi-compartment model, which corresponds to the different tissues of the body that are connected by the circulating blood system figure 3. Each compartment is defined by the tissue volume and tissue blood flow rate that is specific for the species of interest. Therefore for the proper use of the technique essential parameters such as for example the pharmacokinetics, bodyweight and organ weight should be available for input in the model. A database with such physiological parameters can be found for humans and/or animals on the web or can also be found in the literature. The physiological parameters can be obtained from in-vitro, in-silico or in-vivo studies. Representative PBPK models include the in vitro determination for the estimation of ADME (iDEATM) model and the compartmentalized absorption and transit (CAT) model. The technique relies in general on the input of species specific physiological parameters and compound specific information (as for example metabolic clearance (CL)), to predict the plasma concentration profile of the compound in the species of interest after intravenous (i.v.) or oral administration. As already mentioned all information needed such as the physiological parameters can be gathered from the literature. The model attempts to describe mathematically all relevant physiological processes in order to predict any interaction of the compound with the system. The model can predict the amount of drug distributed to target or non-target 15 organs, which is important in determining the physiological response or side effects of the drug. The performance capability of PBPK modeling in predicting oral absorption in humans has been tested by the iDEATM simulation system. The fraction of the dose absorbed to the portal vein (FDp) for some compounds have been predicted by the model and compared to the FDp values obtained from human clinical trials (Figure 4) 6. Plotting of the values against each showed a correlation between the predicted values and the human values, validating the performance of such a model. BPK modeling is a promising tool for the prediction of the behavior of the drug in the body after exposure. There are many reports including the report of the iDEATM simulation system that demonstrates the prediction of PK profiles of drugs in humans by PBPK model. However validation of such simulations for some compounds is difficult when the in-vivo evidence is lacking 2. Therefore introducing the microdosing approach will help fill the gap of the in-vivo data. Combining the microdosing approach and the PBPK modeling technique will therefore lead to a better understanding of the behaviour of a candidate drug during the development process Figure 5. The in-vivo evidence for the verification of the PBPK model simulation can then be obtained from the microdosing study. On the other hand PBPK modeling technique can be also used to analyze the problems that microdosing can encounter when predicting PK values. For example saturation due to binding of the compound to proteins is less likely occur with a microdose, indicating that the PK values cannot be predicted accurately. Hereby a more accurate prediction of PK profiles of a drug or test compound at therapeutic dose in humans can still be obtained from PBPK model analysis based on the microdose clinical study in combination with in-vitro study with human materials 2. Figure 5: Method that includes both the microdosing approach and the PBPK-model analysis for a more accurate prediction of PK profiles of a new drug candidate in human. 16 The PK profiles of a compound can be studied by evaluating the ADME properties of the compound. For the evaluation of these properties there are several methods available, however only a few will be shortly discussed. Evaluating these properties can help to predict the outcome of the microdose study. The absorption properties of a compound can be obtained from in vitro study with human colon adenocarcinoma (Caco-2) cells 27. These cells have similar characteristics as the intestinal enterocytes, such as formation of microvilli, tight junctions, P-glycoprotein (P-gp) and other transporter expression 28. Several studies have found good correlation between Caco-2 permeability and the oral drug absorption in humans. The method consists of measuring the amount of compound that is transported from the medium to the cells. The data is then expressed as the apparent permeability coefficient (Papp) with the unit cm∙sec1, using the following equation: P app = (area × initial concentration × time)⁄ amount transported The difference between the transports of the compound can also be studied by using specific inhibitors. For example the role of P-gp in the uptake of the compound can be evaluated by using Verapamil (inhibitor of P-gp). The presence of the inhibitor would inhibit the efflux from the cells and therefore would enhance the Papp of the compound. The outcome of the result can determine if it is worth to be further studied in a microdose study or not. For example if the uptake of the compound is mostly influenced by the P-gp, indicating that the compound is poorly absorbed through the intestine, it has no significance of performing a microdose study especially not through oral exposure. Binding of compounds to proteins or tissues can influence their distribution across the body. Plasma protein binding can be studied with the methods ultrafiltration and equilibrium dialysis, from which the equilibrium dialysis is the most used method 28. This method is most used because it is not affected by nonspecific binding. However it should be pointed out that it is unsuitable for compounds that are known to be unstable in plasma due to the long time (approximately 20 hours) needed for the system to reach equilibrium. The percentage of plasma protein binding can then be derived from the measured amount of compound remaining in the plasma (Cpl) and the amount of compound found in the buffer (Cbu), with the equation percentage plasma protein binding = 100 × (Cpl − Cbu)⁄ Cpl According to the plasma protein binding percentage a compound can be categorized as highly, moderately or poorly protein bound. Only compounds that show more than 95% binding to proteins is considered highly protein bound. However protein binding interactions have little clinical implications unless the drug has a narrow therapeutic index and is also highly protein bound 28. Nowadays most of the drugs approved have high therapeutic indices. The bioavailability of a compound can also be influences by metabolism of the compound. After oral exposure the drug is absorbed in the intestine and transported through the circulation system to the liver, where it is subjected to the hepatic metabolism followed by the elimination of the compound through the bile or urine via the kidneys. The metabolism process can be divided in two phases: 1) phase 1 (oxidation, reduction or hydrolysis step) that makes the compounds more hydrophilic, and 2) phase 2 (conjugation step) that makes the compounds inactive. Enzymes responsible for the metabolism process are present in the hepatocytes, therefore incubating these cells with the compound followed by measurement of the amount of compound present in the buffer (Cafter incubation) should determine the metabolic stability of the compound 27. In vitro systems that can be used for this study can consist of hepatocytes or microsomes. Microsomes are usually used to determine P450-mediated metabolism (phase 1), however hepatocytes being intact cells are believed to be similar model of the liver and therefore should deliver better drug clearance measurements 29. The metabolic stability is generally expressed as the percentage of parent compound that disappeared and can be calculated with the equation percentage compound disappeared = 100 × (1− (Cafter incubation⁄ Cbefore incubation)) The ability of the liver to metabolize and therefore help eliminate the compound can be determined by estimating the intrinsic hepatic clearance. The apparent intrinsic clearance, the following equation is used Clint,app = (0.693⁄ in vitro t1/2)( incubation volume⁄ mg microsomal protein)(45 mg microsomal protein/ g of liver)(*20 g of liver/ kg bodyweight), where * refers to the values depending on the selected species. Human values are given in the equation. The apparent intrinsic clearance is then corrected for the free fraction of the incubated compound to determine the intrinsic clearance 29. The amount of parent compound remaining also known as the free fraction can be calculated with the equation Fu,mic = (concentration at receiving side×1.5)⁄(concentration at donor side×3) 17 The in vitro apparent intrinsic clearance (CLint) of the compounds by the liver can be determined from the enzyme kinetic data by calculating the ratio of Vmax⁄ Km, known as the maximal rate (Vmax) at which metabolism occurs and the michaelis constant (Km) which indicates the concentration substrate needed to fill half the active sites of the enzyme thus yielding half the maximal velocity (Vmax). By observing the formation of a specific metabolite at constant enzyme concentration exposed by different concentration of compound both the Vmax and the Km properties can be estimated (fig 6). At saturation rate the amount of compound metabolized per unit time can be calculated by [S]×Vmax⁄ Km+[S] After determination of the kinetic properties in vitro, the data can be extrapolated to determine the in vivo situation. As can be observed in figure 6 the plotted data does not show a linear curve, however this can be linearized by using the lineweaver burk method. Hereby 1/reaction velocity is plotted against the 1/ substrate concentration, resulting in a straight line where the slope is defined as Km/ Vmax also known as the CL. Figure 6: Michaelis menten kinetics 30. Plot of the reaction velocity (V0) as function of the substrate concentration [S]. Figure 7: conversion of michaelis menten kinetics plot using the lineweaver burk method 31. As can be imagined from the plot at therapeutic dose the maximal reaction velocity is mostly reached indicating saturation of the system, which is not the case for a microdose. Therefore it can be assumed that in a microdose study the compound would be mostly metabolized, indicating that no clinical effect would be obtained from the parent compound. This also points out how safe a microdose study is. 18 Pediatric drug development As already mentioned in the introduction, the development process for drugs consists of in-vitro and in-vivo studies to obtain toxicity data of the studied compound. The obtained data from experiments with laboratory animals is then extrapolated to the human population to predict the effect of the compound in the human subject. During extrapolation the variation between species and within species are taken into considerations, including the differences between adults and children. Therefore correcting for the variations during extrapolation is expected to correct for the differences between children and adults, indicating that the compound should be considered safe for both adults and children. This however can always be questioned, because most of the toxicological data obtained are from studies with adult animals. Information from studies with juvenile animals would be more relevant to children. On the other hand this could also lead to great uncertainties for the risk assessment in children due to the different maturation rates of several systems such as the enzyme activity, which may affect the pharmacokinetics of a compound between the species. Maturation of the enzyme system for example is known to start in the first 2-3 months in humans, whereas in rats it occurs in the first 2-3 weeks of life 10. Generally dosage of the compounds is mostly determined for the adult human. For children the dosage is therefore corrected for bodyweight and/or body surface area in order to be more indicative. The most preferred method for prescribing therapeutic dosage of drugs in children is by correcting for body surface area instead of correcting for bodyweight. Correction for the body surface is expected to give better adjustment parameters, because it describes the total body water and extracellular water better 10. Therefore dosing based on correction for the surface area may avoid underdosing of water-soluble drugs. Another advantage is that both the measures of height and bodyweight are required to estimate the size. This indicates that most variables are considered in the correction of adult dosage to a proper dosage in children. Correction of the adult dosage to a more proper dosage in children however does not indicate that the dosage is good for children of different ages. Children of different ages can show great differences in for example the absorption of drugs, indicating that the corrected proper dosage is more indicative in children that have equal PK properties as the adults. Variability of pharmacokinetics can be observed in both children and adults. However PK variability can also be observed within the group of children, due to the grouping of different ages (e.g. 1 week or 1 year of age). Variability in pharmacokinetics of children is shown in figure 6. Neonates and infants are shown to have great variability, indicating that the corrected dosage can lead to an overdosing or an underdosing with the related adverse effect in children of this age. The variability differences is of great importance for the risk assessment in children 11. Table 6: Children classification according to age. Term Neonates Infants Age (month) 0–1 Figure 7: Child/adult half life ratios across drugs and age groups 11. Plotting the half life ratio of different compounds for both children and adults according to their age in days gives an indication of the variations according to age. 1 -12 Toddlers 12 – 36 Children 36 – 120 Adolescent 120 - 228 19 Either overdosing or underdosing of a drug in children indicates that the efficiency of a drug is unknown, resulting in different toxicity effects in children of different ages. This can be mostly explained by pharmacokinetic and pharmacodynamic differences in children. As an example the drug acetaminophen or better known as paracetamol is taken. Adults are more sensitive to an overdose of paracetamol than children. In adults paracetamol is normally eliminated from the body through glucuronidation, however the glucuronidation level observed in children are low indicating that children have other elimination routes compared to adults. Higher rate of glutathione turnover and also more active sulfate conjugation is observed in children, which compensates for the low level of glucuronidation resulting in a faster elimination of the active compound 12. This example illustrate that there are important differences between children and adults that need to be consider in order to obtain a therapeutic dosage for children or for the development of new drugs, besides only the adult data obtained from experimental studies. Differences in growth or maturation of specific systems that may have an impact on the absorption, distribution, metabolism and elimination of a compound may explain the observed variability in pharmacokinetics. 20 Differences in children vs adults The growth and development process of different organs of either animals or humans occur at different rates, indicating that differences in pharmacokinetics and toxicokinetics between the various age groups is most likely to occur. Therefore the differences should be taken into consideration for the development of generic drugs. The main physiological differences observed in human adults and children have been well studied during the years and are summarized in table 7. These physiological differences, such as the immature gut development and immature enzyme activity, can lead to different PK properties of a drug according to age of the treated patient. Researchers have studied the effect of age on the pharmacokinetics in humans both invivo and in-vitro, and concluded that the most dramatic physiological changes that may affect the pharmacokinetics of a drug occur in the first 6-12 months 10. Research for the physiological changes in animals from different ages on the other hand is minimal. The pharmacokinetics is mostly affected by changes in the absorption, distribution, metabolism and elimination process of compounds, better known as the ADME. Table 7: Summary of developmental changes affecting pharmacokinetic Physiological system Gastrointestinal tract Body compartments Plasma protein binding Metabolizing enzyme capacity Renal excretion Observation in children Immature gut, less surface for the absorption of compounds. Low P-glycoprotein expression. Decreased fat, decreased muscle mass, increased total body water. Limited serum protein binding capacity. Immature enzyme function Decreased glomerular filtration rate and active tubular function Pharmacokinetic implication Slower uptake of compounds. Without affecting the bioavailability. For some compounds such as metals, an increased uptake is observed. Low P-glycoprotein results in less excretion and thus more absorption of compounds. Less distribution and retention of lipid soluble chemicals, whereas the water soluble chemicals have larger volume of distribution (Vd). More free toxicants in the blood circulation. Slower metabolic clearance, less metabolic activation or elimination of activated metabolites. Reduced plasma clearance and increased elimination half-life of compounds Absorption The primarily route of exposure for most medications is through oral ingestion and therefore the absorption of drugs by the gastrointestinal tract is of great importance. The major function of the small intestine is to facilitate the absorption of nutrients and compounds. Children have relative smaller surface area than adults, indicating that there are less receptors and transport proteins present per square unit intestine and thus will result in slower absorption of compounds. A study with 580 hospitalized children receiving drugs through a feeding tube, showed that the rate of absorption is related to age 10. The absorption was much slower in neonates and young infants than in older children. The absorption of nutrients and ions in the intestine occurs through active transport processes present in the intestine. The expression of these transport processes is in line with the needs of a growing child. Therefore absorption of compounds that involves these transport processes is most likely to be better absorbed in the children. A classic example to illustrate this is the absorption of lead. The default oral absorption factor used by USEPA’s biokinetic model for lead uptake in children is defined as 50% for lead in diet/drinking water and 30% for lead in soil/ house dust. However the absorption of lead in the human adult gastrointestinal tract only accounts for 10%. The great difference in lead absorption between adult and children is partially attributed to the greater pinocytic activity of the intestinal epithelium in young children before closure or maturation of the 21 gastrointestinal tract 11. However there are also evidences that suggest that the presence of milk in the gastrointestinal tract may promote the absorption of metals in neonates. One of the many transporters that plays an important role in the absorption of xenobiotics, are the transporters belonging to the ATP binding cassette (ABC) superfamily such as the P-glycoprotein. The P-glycoprotein has been detected in the apical surface of epithelial cells of excretory organs, suggesting its protective role against xenotoxins. The Pglycoprotein can protect the host by enhancing the excretion of these compounds or preventing their uptake from the gut. The presence of the transporter is already detected in the early human fetal life (11-14 weeks), however this is far not the adult levels. The expression of P-glycoprotein in mice was reported to be low at birth and increased significantly with the maturation of the intestine. Distribution After absorption the compound is mainly distributed in the body according to its chemical properties, which indicates that the body composition of a subject plays an important role in the distribution of chemicals within the body. Newborns and infants have different body composition of water and lipid compared to older children and adults. At birth there is more body water (expressed as percentage of total bodyweight) than in adults, e.g. 78% in neonates and 55% in adults. The total body water of children reaches the adult level by 12-years of age 10. Besides the high body water and thus low lipid body content, the adipose tissue consists also partially of water. The adipose tissue of infants consists of 57% water and 35% lipids, whereas the values in adults are 26% and 71% respectively. Therefore in general the lipid soluble compounds are less likely to distribute through the body at birth, whereas the water soluble compounds on the other hand will have a large volume of distribution (Vd). Body lipids of children will increase after birth in its first 9 months of life, and then will decrease again until the ages 4-7 years. Hereafter it will increase again reaching adult levels. This illustrates that the distribution of lipid soluble substances and therefore the half-life of the lipid compounds can change during the development process and growth of children. Besides the body composition there are also other factors that can influence the distribution of compounds. The binding capacity of plasma proteins for example can affect the distribution of compounds. Binding of the compounds to plasma proteins has two consequences, which are: 1) a slow elimination process of the compound and 2) that the compound is not free to exert its effect. Only free compounds can exert effect to target cells, indicating that binding of the compounds to proteins limits the amount of free compounds. The measured amount of total plasma protein concentrations in neonates is approximately 59 g/L, whereas in adults it is approximately 72 g/L. At first the difference in concentration does not seem so dramatic, but due to different binding capacities of proteins it can have a great impact. The protein binding capacities of neonates is low regarding both albumin and alpha-1-glycoprotein, indicating that relative high levels of circulating free active drug can be found in children. Metabolism Clearance of a drug usually starts with metabolism. The primary organ responsible for the elimination of compounds through metabolism is the liver. For the metabolism process however maturation of enzymes involved in the process is needed. Maturation of the enzyme system starts in humans in the first 2-3 months, whereas in rats it occurs in the first 2-3 weeks of life. Observations in infants showed that some drugs tend to have longer half-life (t1/2) and lower clearance, indicating the metabolism capacity of children. The enzymes activity responsible for metabolism will increase after birth until it reach adult levels by 1-3 years of age 10. Metabolism in general is divided in two steps: 1) phase 1, and 2) phase 2. Phase 1 involves mostly oxidation, hydrolysis and reduction of a compound, whereas in phase 2 the compound is conjugated with for example glutathione making it more water soluble and thereby increases its elimination from the body. The Phase 1 metabolism step consists mainly of the cytochrome P450 enzyme system. The CYP P450 enzymes are shown to remain stable from the first trimester of gestation till 1 year of age, and accounts for 30- 50% of the adult level. The enzyme levels in neonates and infants ranged from 0.08 to 0.13 nmol/mg protein, whereas in adults levels of 0.3 ± 0.037 nmol/mg protein is found 10. Several CYP’s, such as CYP1A2 and CYP2E1 are present at low level at birth and increases hereafter rapidly in the first year of life. The low levels of CYP1A2 at birth is also supported by in-vitro studies 11. Theophylinne is compound that is 22 primarily metabolized by CYP1A2. The metabolic clearance of theophylline is low in neonates and infants with a limited metabolizing capacity and therefore it is mostly excreted as the parent compound itself. The level of the CYP1A2 enzyme is shown to approach adult level by 2-6 months of age. Expression of the enzyme CYP2E1 is also shown to be low at birth, however in contrast to CYP1A2 it increases its level rapidly during the first 24hr after birth achieving adult levels in children at 1-10 years of age. Another enzyme of the CYP P450 family is the CYP3A4 enzyme. This enzyme is the most abundant enzyme expressed in adults and is known to be responsible for the metabolism of allot of chemicals. In neonates however the enzyme is found in very low levels. The mRNA levels of the enzymes in the foetal liver microsomes accounted for 10% of adult levels. After birth the levels of the enzyme increases rapidly reaching 50% of adult levels at 6-12 months of age 10. Surprisingly there are also studies that show that CYP3A4 activity in children may exceed the adult levels at the second months until the first 2-3 years of life. Another enzyme belonging to the CYP3A subfamily is the CYP3A7. In contrast to the other enzymes, the enzyme activity level of CYP3A7 is higher in newborns relative to adults. Although both CYP3A7 and CYP3A4 have similar substrate specificities, they are shown to have different functional activity. Midazolam is metabolized by CYP3A4, suggesting that the compound may also interact with the CYP3A7 enzyme due to the similar substrate specificity. The compound is shown to be slightly metabolized by CYP3A7 in neonates, resulting in a reduced clearance of the compound. This also support the low CYP3A4 activity present at birth 10. Despite the high levels of CYP3A7 present in newborns, it is still not clear what the metabolizing capacity of the enzyme is. Enzymes responsible for the phase 2 metabolizing step are among others the uridine glucuronosyl transferases (UGT), glutathione S-transferases (GST) and sulfotransferases. UGT is in general responsible for the glucuronidation of most hydrophobic compounds making them more hydrophilic and therefore enhances it elimination from the body. Studies have shown that neonates and infants have limited glucuronidation capacity compared to adults. This could be illustrated by studying the elimination of the compound acetaminophen, which is mainly eliminated through glucuronidation. Children should be expected to have a reduced excretion, because of their reduced glucuronidation activity. Despite the reduced enzyme activity, paracetamol is yet eliminated as a sulfate conjugate in children. Until 9 years of age the sulfation capacity in children is increased, which compensates for the limited glucuronidation activity enhancing the elimination process of compounds such as for example paracetamol 11. This indicates that compounds may be cleared in early life stages by alternative pathways. The enzyme glutathione S-transferases (GST) play an important role in the detoxification of electrophillic alkylating agents 10. The ability of glutathione conjugation is already present in utero and at birth thanks to the dominant presence of the GST-pi (foetal) isoform 11. Other isoforms such as GST-mu and GST-alpha are also present but in much lower levels compared to adults. The presence of high level of GST-pi in the early life stages does not indicate that its ability to detoxify all compounds is efficiently. The substrate specificity of the different isoforms is not well known, which suggests that it is unknown if the substrates that are specific for GST-mu may be detoxified by the GST-pi or not. A study showed that human fetal liver cytosol was 33% less active compared to adults. The conjugation of glutathione with benzo(a)pyrene-4,5-oxide was lower compared to adults 11. Another enzyme responsible for the phase 2 metabolizing process is glutathione Stransferases (GST). The ability of GST to detoxify compounds by conjugation with glutathione depends of the amount of glutathione (GSH) available. GSH can be depleted, which means less GSH available and therefore subjects are more susceptible to the toxic effects of compound. Plasma GSH levels in infants increases from 1µM during the first week of life to approximately 3µM at 1 year of age, which is similar to the adult levels of 34µM 11. Besides the enzymes present in the liver, there is another factor that plays an important role in the metabolism process of compounds in the intestinal flora. The bacterial flora of the gut lumen varies according to the different ages of life, and is also capable of metabolizing compounds. The enterobacteria and enterococci are the dominant bacteria present at birth, however after 7 days of birth the bifidobacteria increases becoming at 13 days of birth the dominant bacteria. The change in microflora is accomplished by the ingestion of fresh milk, which will promote the growth of the bifidobacteria. The food intake 23 of neonate’s changes according to their development and growth enhancing further changes of the intestinal flora by decreasing the presence of lactobacilli and increasing the putrefactive bacteria. The change in bacterial flora is important for the hydrolosis of compounds that are conjugated and secreted in the bile. Absorption of unconjugated compounds by the intestinal epithelium is facilitated by changes in the microflora 10. Besides the bacteria there are also another enzyme namely ß-Glucuronidase, responsible for the metabolizing capacity in the gut. ß-Glucuronidase is an enzyme responsible for the unconjugation of glucuronidated compounds. This enzyme is known to be absent in the human adult gut, whereas in children it can be detected. This indicates that the neonatal gut is capable of converting glucuronides to their unconjugated state which can then be enterohepatically reabsorbed. In general it can be concluded that the ability of children to metabolize certain compounds in the gut depends on the maturity of the bacterial enzyme system present in the gut lumen. Elimination Excretion of compounds occurs through the bile into the faeces or through the kidneys into the urine. Renal excretion is for most compounds the predominant step in the elimination process. The excretion capacity of the kidneys is reflected by the clearance rate and half-life of compounds. Neonates are shown to have high bodyweight-normalized clearance compared to adults, indicating their fast elimination rate. The elimination rate depends on the amount of compound that is filtered per unit time defined as the glomerular filtration rate. This can be influenced by the renal plasma flow and binding of compound to proteins. The kidneys of neonates are known to receive only 5-6% of the cardiac output compared with 15-25% for adults, resulting in a renal plasma flow average of 12ml/min at birth and 140ml/min in adults which is achieved by 1 year of age 10. A reduced renal plasma flow also suggests a reduced glomerular filtration rate (GFR). In neonates the GFR is found between 8 and 20ml/min that then immediately increases achieving the adult levels at 1.56 months of age. Besides this factor the proximal tubules of the kidney in infants are small, indicating that there are less functional tubular cells present which leads to a reduced transporter (secretory) system. The tubular function present in neonates is established to achieve the adult level at 30 weeks of life after correction for the body surface area. By using the tubular transport maximum for para-amino hippurate (TmPAH) as an indicator for tubular function, it could be observed that in the first year of life a 10fold increase in TmPAH occurs. Another important transporter is the organic anion transporter (OAT) family, which removes anionic substances from the blood via a transport mechanism present in the basolateral membrane of renal epithelial cells. OAT-gene expression in rats has been shown to increase through birth, following a decrease after birth to reach adult levels 10. Many drugs such as penicillin are excreted by the transport system present in the proximal tubules. This indicates that the deficiency in the system may cause longer half-lives for the compounds. 24 Dosage and effects of drugs in children Since 1997 there have been new rules related to the pediatric drug development due to the variability in both the efficiency and toxicity potency of drugs between adults and children. In 2007 the pediatric research equity act (PREA) has been reauthorized, which describes the practice of pediatric clinical studies of certain drugs to obtain pediatric labeling for new indications, new dosage forms, new route of administration, new dosage regimens, and/or new active ingredients 12. Indicating that candidate drugs that passed the clinical trials during the drug development process and are therefore approved for the market, can be tested in a pediatric setting to obtain information relative to its use in children. Hence for the development of pediatric drugs more studies are needed, including time and the participation of more patients, which eventually will result in increased developmental costs. The human investigational ethical assurance has kind of impeded the participation of children in clinical trials with elements as “respect for persons” and “human autonomy leading to concepts of voluntary informed consent” 12. This makes pediatric clinical trials challenging and therefore a proper balance between scientific, ethical and practical concerns is required. One of the most challenging drug trials are those with analgesic such as paracetamol, because during the trial subjects are most likely to experience pain for an extended period of time 13. The most common pain relievers used in the human population are among others paracetamol, diclofenac and sumatriptan. The mechanism of action and effects of these compounds are in general well known in adults, especially for paracetamol. However the efficacy of the drug in children depending on their age is mostly unknown and requires more studies. As already mentioned in section Differences in children vs adults, maturation of the enzymes involved in the metabolism process reaches adult levels at approximately 3 years old of age. Indicating that extrapolation of adult data to predict efficacy of drug in children is justified, due to equal metabolic clearance of the compound between children and adults. For children younger than 3 years of age the efficiency and toxicity data of compounds are needed for the proper selection and dosage of the medicated drug. Overall conclusion is that there are more studies needed to elicit the pharmacokinetic and pharmacodynamic properties of a drug in children. Determining these properties will result in the selection of the proper drug for treatment and the proper dosage according to the age of the subject. Prescriptions of drugs to children are preferably based on the available information from studies with young subjects. However in the absence of such information the prescription of medicines relies on the efficiency and toxicity data obtained from experiments with adult subjects. In general this indicates that a proper dosage for the young generation is not known. Paracetamol is a general safe and well tolerated analgesic and antipyretic drug that is frequently prescribed in children. In general, the dosage of paracetamol in children is based on a single dose of 10 mg per kg bodyweight which can be repeated every 4-6 hours. Despite the general dosage children are frequently being overdosed (young children) or underdosed (older children) together with the associated toxicity risk or risk for under treatment 14. In the emergency room pediatric patients are commonly exposed to paracetamol for the reduction of pain. The prescription is mostly based on the recommendations in books, which also includes the current best practice based solely on experience rather than on scientific basis 15. Many of the recommendations involve the use of unlicensed or off label medicines. Unlicensed medicines are used as an alternative for the lack of licensed medicines. Off label is defined as medicines used without available pediatric information or contraindication in children or that the drug is used different as is indicated in the literature 15, 17. A study in a Spanish emergency room showed that 70% of the drugs administered to pediatric patients were in an off-label manner 17. A total daily dose of 90 mg/kg per day for paracetamol either orally or rectally for 48hr is recommended in order to provide an adequate analgesic level of the drug and thus relieve the patient of postoperative pain. 30% of all off label prescription is related to the prescription of paracetamol, from which 20% is prescribed to children resulting in high or 25 low dosage exposure 14. Prescription of paracetamol to children is mostly based on the concerns of potential hepatotoxiciy (observed at doses of 150mg/kg per day and above) rather than the pk properties of the drug. In the literature there are PK properties for paracetamol in children reported, however it is only valid for ≥5 years old children. Little information is available for neonates and infants. A clinical study involving children of 5-15 years of age having a tonsillectomy, determined the pk properties of paracetamol. The children were orally exposed to 22.5 mg/kg paracetamol after they had the surgery in order to determine the efficacy of the drug in the reduction of pain. The obtained pk properties were T1/2 = 15H, Cmax = 12.7 µg/ml, V=1.06 L/kg and CL = 0.68 L/H/kg, which is in concordance to the properties found in the literature for adults (T1/2 = 2H, Cmax = 0.02 ng/ml, V=1.0 L/kg and CL = 0.3 L/H/kg) with the exception of T1/2 and Cmax 18. Another pediatric study investigated the pharmacokinetic changes in children of different ages. The study showed that the total body of clearance for paracetamol increases whereas the volume of distribution decreases after birth. Clearance of a newborn is estimated to be 62%, whereas the volume of distribution is 174% of older children. Clearance of paracetamol is estimated to be 0.15 L/H/kg in neonates and 0.37 L/H/kg in infants. The study also estimated that for reaching target concentration higher than 10 mg/L exposures of 45 mg/kg per day is needed at birth, whereas for 5 and 8 years old children the exposure needed are 90 mg/kg and 75 mg/kg per day respectively 19. Target concentration of 10 mg/L is defined as the steady-state concentration needs to reduce 50% of the maximum possible temperature observed in febrile children and is also the effect site concentration associated with pain scores less than 6/10 in children after undergoing for example a tonsillectomy. The pharmacokinetics of a drug is very important to determine the proper therapeutic dosage for a drug to elicit its effect. The effects of a drug can be further explained by studying the mechanism of action of the drug and the pharmacodynamic properties. The pharmacodynamics of a compound should elicit how the compound is distributed around the body of the subject and therefore determine in which compartment it mostly exerts its effect. The mechanism of action of paracetamol for instance is well studied. However due to the results it is still debated and elusive, indicating that the precise mechanism of action is still not known. Among the different mechanism of action it is thought that paracetamol is metabolized into an active metabolite after it crosses the blood-brain barier and thus enters the central nervous system (CNS). Concentrations of 1.3–18 mg/L have been detected in the cerebrospinal fluid of children undergoing surgery after a single intravenous injection of 15 mg/kg paracetamol, whereas the plasma concentrations ranged between 2.4 and 33 mg/L 20. The active metabolite will then promote endogenous cannabinoid activity that results in a prostaglandin-mediated cyclo-oxygenase enzyme 5 (COX-5) effect 21. The analgesic capacity of paracetamol is associated with the availability of cannabinoid receptors in the nervous system. Blockage of the cannabinoid CB1 receptor prevents the analgesic effect of paracetamol 22. Further mechanism for the analgesic capacity of paracetamol includes the suppression of the signal transduction from the dorsal superficial layers to the spinal cord. Metabolites of paracetamol e.g. NAPQI, act on the TRPA1-receptor in the spinal cord inhibiting the signal transduction pathway 16. Besides the ability of paracetamol to reduce fever and pain in the treated patient, it can also induce adverse effects. A study showed the use of paracetamol in the first year of life is associated with a dose-dependent increased risk of asthma symptoms 23. Medium and high use of the paracetamol resulted in an odss ratio (OR) of 1.61 [95% confidence interval: 1.36-1.56], whereas no use resulted in OR 3.23 [95%-CI: 2.91-3.60]. The calculated population-attributable risks rang between 22% and 38%, making paracetamol a risk factor for the development of asthma in children. 26 Pediatric microdosing Children are mostly treated by drugs that, although its usage is approved in adults, it is not yet approved for use in this group of the population. The unapproved usage of these drugs in children is mainly due to incomplete labeling guidelines. This can lead to therapeutic incidences, such as for example the incidence with the drug thalidomide. In the 1960s several children were born with stunted arms or legs due to exposure of the mother to thalidomide for the treatment of morning sickness during the pregnancy 12. This disaster was the leading cause that triggered the regulatory affairs to facilitate the examination of drugs in this sensitive group of the population. The involvement of children in clinical trials has increased by approximately 1.5 fold between the years 2000 and 2006 12. Pharmaceutical industries are paying to obtain pediatric data, however the barrier of obtaining the consent of parents to use a drug in an off label manner on their children (especially in studies using a placebo) still persists. Although a clinical study is a wellcontrolled study, not knowing what will happen to their children after exposing them to such as drug certainly disturbs them. A microdose on the other hand is known to be harmless because of the low dose used in the study. However this does not imply that parents will give their consent easier. It is still their child and depending on their interest in science will give permission or not. Performing a pediatric microdose study will give information about for example the pharmacokinetics in children, which then can be used to determine if further studies for that compound are worth or not. If data from the microdose study demonstrates that the compound is easily cleared from the body or maybe not absorbed then it is not worth starting a clinical phase 1 study without changing the exposure properties of the compound. Therefore a microdose study can determine if the compound that is approved for usage in adults can be used for treating children. Traditionally the dosage of drugs for children is obtained by correcting the dosage of the drug in adults by the bodyweight and/ or body surface area such as mentioned in the chapter pediatric drug development. However this can lead to an underexposure or overexposure of children to the drug, depending on the physical properties of the children for example children with obesitas. On the other hand correction for bodyweight and/or body surface area does not consider the pharmacokinetic differences between adults and children. As summarized in the chapter differences in childern vs adults, children have for example a reduced amount of P-glycoprotein compared to adults, which indicates that excretion of compound through this pathway will be reduced in children meaning that the amount of drug in the blood circulation will be elevated compared to the level observed in adults. The absolute bioavailability for the compound can be determined from the microdose study, which is eventually the dose that exerts the effect eventually. From a microdose study also the Cmax and Tmax for the compound can be determined, referred as maximum concentration and time elapsed to reach maximum concentration. This information can then be included for example a PBPK model to determine the effect or fate of the compound in children. By running simulations of children of different ages exposed to the dose normalized for the weight obtained from adult data, the exposure of the cells or organ compartments can be predicted. Determining therefore if age-specific dose changes are necessary to achieve a relatively constant drug exposure in adults and children 32. 27 Conclusion Microdosing is a promising approach that helps reduce the failures of drugs during the developmental process and therefore also reduces the high developmental costs. More drug candidates could be tested for the same developmental cost, indicating that a great variety of drugs may be placed in the market. Therefore more drug variations are available for patients in case of adverse effects due to ingredient present in the formulation of the medicine. This is particularly the case with allergic reactions to certain compounds. Besides for the development of drugs in adults, microdosing could also be used to predict PK properties and proper dosage of drugs in children. It is extreme difficult to start a clinical trial with children because of the ethical implications. However with such a low dose that is considered to be safe, it should be possible to start a clinical trial with children. This would help in the development of proper drugs for children. Drugs with better PK properties in children should be more efficient for treatment. Determination of the PK properties of a compound in children can also lead to proper estimation of the therapeutic dosage. These properties can then be included in the PBPK modeling technique for simulation of the weight-normalized dosage obtained from adult data to determine the exposure of the different organ compartments. From results it can be estimated whether correction for age-specific dose changes should be performed in order to obtain the same constant drug exposure as observed in adults. 28 Discussion Before a microdose study can be carried out, the candidate drug should pass a variety of test to assure the safety of the subjects that will be exposed to the compound. One of the tests describes the establishment of the NOAEL and MTD in one animal species. This partially contradicts as mentioned in the introduction the aim of the microdosing approach in reducing the amount of animals needed for testing of the compounds in an experimental setting. Despite the extreme concern of safety, it should be noticed that the regulatory agencies defined a microdose to be a 100th of the predicted pharmacological dose or 100µg. Therefore it could be questioned whether the amount of drug needed for the test can be calculated by applying the standard toxicological safety factor of 100, after considering the weight of the subjects 1, 15. This would result in less animal studies needed before a microdose study can take place. The microdosing approach looks very promising for the development of new drugs for the children. Because the NOEL and MTD is determined before a microdose study can occur, it can be considered to be safe to be tested in children. Allot of drugs that are being administered to children has never been tested on children. The dosage of such drugs is mostly based on correction of the therapeutic adult dose for the bodyweight and bodysurface area. In this case a microdose study could help determine the proper dose for children. The pharmacokinetics of drugs in human is shown to vary according to the age of the subject. Great variability is shown within the group of children, namely in infants. Therefore dosage of drugs corrected from adult therapeutic dose could lead to underdosing or overdosing in combination with its related effect in children. Microdosing in combination with PBPK modeling technique could determine the behavior of the drugs after the children are exposed, gaining more information about the dose-effect relationship based on scientific evidence rather than only the results observed from years of experience. Correcting of the therapeutic adult dose for dosing in children could lead to problems in the case of obese children. The actual bodyweight of obese children may be in the range of the adult bodyweight, indicating that these children may receive doses outside the allowed dosing regimen for children of the same age. This is likely to occur in drugs that are already licensed as for example with paracetamol. The amount of paracetamol used to relieve pain in children after having an operation procedure is extremely high, compared to the dose prescribed in the label of the drug. As already mentioned children are less sensitive to a paracetamol overdose compared to adult, due to their rapid ability to eliminate the compound. 90% of premature babies in a healthcare setting are exposed to drugs that are either unlicensed or used in an off label manner. Unlicensed drugs are used as an alternative when no commercially produced licensed drug is available 15. This is extreme dangerous due to the lack of information about the dose-effect relationship and adverse effects in babies. Children have always been considered as the most delicate and vulnerable group for toxic effects. However they are more likely to be exposed to an overdose of drugs in an acute or chronic manner depending on the treatment and thus are also exposed to its toxic effect. For a proper risk assessment it should be determined if the internal exposure of children is within the drug’s therapeutic window, or if it is more likely to exceed the NOAEL of the compound 10. The toxicological data obtained from animal studies used to determine the dosing regimen in humans is obtained from studies conducted on adult animals. As in the case of humans the physiological aspects can also differ in animals of different ages. Indicating that the observed values in adults is not necessary the same for the younger animals. Research for the physiological changes in animals from different ages is minimal, whereas in humans it is well studied. Traditionally during the development process of drugs, all compounds are first tested in animals before it can be tested in a clinical setting on humans. Therefore dosing of animals in the first days of life would give more information about the sensitivities in children due to their immature ADME-relevant proteins. From the studies also the toxicological data such as the effective dose concentration (EC50), NOAEL and margin of safety can be determined. When extrapolating these data obtained from animal studies to predict the values in the human population it is important to note the slope of the dose-response curve. The slope can be linear or curvilinear and 29 determines the toxicity of the compound. The NOAEL obtained from the studies is then used to calculate the acceptable daily intake of the drug and also to calculate the reference dose (RfD) for chronic oral exposure. This is achieved by dividing the NOAEL by uncertainty factors (UF) and modifying factors (MF) from the selected study for a specific critical endpoint. RfD = NOAEL / (UF x MF) 4, 24. A problem associated with using the NOAEL to determine the RfD is that the dose must be a dose that has been tested during the experimental phase and resulted in no observed effect. This indicates that the shape of the dose-response curve in the lower part of the curve is ignored and unknown. The benchmark dose (BMD) approach can in that case help resolve the problem. The BMD approach models the dose-response curve by the software, from which the 95%confidence interval of a dose associated to a benchmark response (BMR) level is calculated. The BMR is usually 1-10%, and therefore it indicates the dose associated with a low level of risk, such as 1-10% 4, 24. The 95%-confidence interval of the BMD can be divided by the lower range (BMDL) and the upper range (BMDU) of the interval. By looking at the ratio of BMD associated to different BMR, such as for example 5% and 10%, the steepness of slope of the dose-response curve could be established. This could be applied to determine the toxic impact of a compound in a population group. 30 Reference list 1. Lappin G, Garner RC. Innovation: Big physics, small doses: the use of AMS and PET in human microdosing of development drugs. Nature reviews drug discovery. 2003;2:p2338p. 2. Sugiyama Y, Yamashita S. Impact of microdosing clinical study - Why necessary and how useful? Adv Drug Deliv Rev. 2011;63:494-502. 3. R.Colin G. Less is more: the human microdosing concept. Drug Discov Today. 2005;10:449-451. 4. Curtis D. Klaasen, John B. Watkins III. Casarett & Doull's Essentials of Toxicology. the McGraw-Hill companies, Inc.; 2003. 5. Outcomes from EUMAPP- A study comparing in vitro, in silico, microdoe and pharmacological dose pharmacokinetics. . 2007. 6. Grass GM, Sinko PJ. Physiologically-based pharmacokinetic simulation modelling. Adv Drug Deliv Rev. 2002;54:433-451. 7. Wilding IR, Bell JA. Improved early clinical development through human microdosing studies. Drug Discov Today. 2005;10:890-894. 8. Laurence L. Brunton, John S. Lazo, Keith L. Parker. Goodman & Gilman's: The Pharmacological Basis of Therapeutics. 11th ed. ; 2006:1794. 9. Wolfgang A. Ritschel, Gregory L. Kearn. Handbook of Basic Pharmacokinetics Including Clinical Applications. ; 2009:184-423. 10. de Zwart LL, Haenen HEMG, Versantvoort CHM, Wolterink G, van Engelen JGM, Sips AJAM. Role of biokinetics in risk assessment of drugs and chemicals in children. Regulatory Toxicology and Pharmacology. 2004;39:282-309. 31 11. Ginsberg G, Hattis D, Sonawane B. Incorporating pharmacokinetic differences between children and adults in assessing children's risks to environmental toxicants. Toxicol Appl Pharmacol. 2004;198:164-183. 12. Andrew E. Mulberg, Steven A. Silber, John N. van den Anker. Pediatric Drug Development: Concepts and Applications. USA: Hoboken, New Jersey : Wiley-Blackwell; 2009. 13. Berde CB, Walco GA, Krane EJ, et al. Pediatric Analgesic Clinical Trial Designs, Measures, and Extrapolation: Report of an FDA Scientific Workshop. Pediatrics. 2012;129:354-364. 14. Kazouini A, Mohammed BS, Simpson CR, Helms PJ, McLay JS. Paracetamol prescribing in primary care: too little and too much? Br J Clin Pharmacol. 2011;72:500-504. 15. Conroy S, Peden V. Unlicensed and off label analgesic sue in paediatric pain management. Paediatr Anaesth. 2001;11:431. 16. Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, Andersson A, Bucher B, Galzi J-L, Sterner O, Bevan S, Högestätt ED, Zygmunt PM. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9tetrahydrocannabiorcol. Nature Communications 2: 551. 2011. 17. Morales-Capri C, Estan L, Rubio E, Lurbe E, Morales-Olivas FJ. Drug utilization and off-label drug use among Spanish emergency room paediatric patients. Eur. J. Clin. Pharmacol. 2010:315-316, 317, 318, 319, 320. 18. Romsing J, Ostergaard D, Senderovitz T, Drozdziewicz D, Sonne J, Ravn G. Pharmacokinetics of oral diclofenac and acetaminophen in children after surgery. Paediatr Anaesth. 2001;11:205. 19. Anderson BJ, Woollard GA, Holford NHG. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol. 2000;50:125. 32 20. Kumpulainen E, Kokki H, Halonen T, Heikkinen M, Savolainen J, Laisalmi M. Paracetamol (Acetaminophen) Penetrates Readily Into the Cerebrospinal Fluid of Children After Intravenous Administration. Pediatrics. April 2007;119:766-771. 21. Elaine M. W. Systemic analgesics in children. Anaesthesia & Intensive Care Medicine. 2010;11:217-223. 22. Ottani A, Leone S, Sandrini M, Ferrari A, Bertolini A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur J Pharmacol. 2006;531:280-281. 23. Beasley R, Clayton T, Crane J, et al. Association between paracetamol use in infancy and childhood, and risk of asthma, rhinoconjunctivitis, and eczema in children aged 6–7 years: analysis from Phase Three of the ISAAC programme. The Lancet. 2008;372:10391048. 24. Ernest Hodgson. A Textbook: Modern Toxicology. 4th ed. John Wiley & Sons, Inc; 2010. 25. www.ICH.org M3 Safety Guidelines. 26. Kannan Krishnan, Melvin E. Andersen. Quantitative Modeling in Toxicology. WileyBlackwell 2010. 27. Albert P. Li. Screening for human ADME/ Tox drug properties in drug discovery. Drug Discovery Today. 2001 ; 6 (7) : 357-366. 28. Mitchell N. Cayen. Early Drug Development : Strategies and Routes to First-inHuman Trials. Wiley & Sons, Inc. 2010. 29. Chuang Lu, Ping Li, et al. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug metabolism and disposition. 2006; 34 (9): 1600-1605. 33 30. http://www.ncbi.nlm.nih.gov/books/NBK22430/ 31. http://www.graphpad.com/prism/tutorials/lineweaver-burk/linwvrburk.htm 32. Feras Khalil and Stephanie Laer. Physiologically based pharmacokinetic modeling : methodology, applications, and limitations with a focus on its role in pediatric drug development. Journal of biomedicine and biotechnology. 2011 (2011), article ID 907461,. 13 pages. 34