Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Psychopharmacology wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Nicholas A. Peppas wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Drug design wikipedia , lookup
Prescription costs wikipedia , lookup
Drug interaction wikipedia , lookup
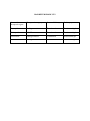
Niosome and Proniosome – Vesicular Structured Dosage Form for Targeted Drug Delivery System Yashavantsinh Chavda1*, Bhavin Bhimani1, Dr. Upendra Patel1, Ghanshyam Patel2, Dhiren Daslaniya2 1. Department of Pharmaceutics, Arihant School of Pharmacy & BRI, Adalaj, Gandhinagar. 2. Department of Pharmacy, JJT University, Jhunjhunu, Rajsthan. Corresponding authorYashavantsinh Chavda Email ID- [email protected] Ph. NO- 09924362838 INTRODUCTION Drug targeting is a specific form of drug delivery where the pharmacological agent is directed selectively to its site of action or absorption. The specific site may be a particular organ, structure, cell or an intracellular region. Targeted drug delivery is a method of delivering medication to a patient in a manner that increases the concentration of the medication in some parts of the body relative to others. Targeted drug delivery seeks to concentrate the medication in the tissues of interest while reducing the relative concentration of the medication in the remaining tissues.1This improves efficacy of the while reducing side effects. Drug targeting is the delivery of drugs to receptors or organs or any other specific part of the body to which one wishes to deliver the drugs exclusively.2 The drug’s therapeutic index, as measured by its pharmacological response and safety, relies in the access and specific introduction of the drug with its candidate receptor, while minimizing its introduction with non-target tissue. The desired differential distribution of drug its targeted delivery would spare the rest of the body and thus significantly reduce the overall toxicity while maintaining its therapeutic benefits. The targeted or site- specific delivery of drugs is indeed a very attractive goal because this provides one of the most potential ways to improve the therapeutic index of the drugs.3 Targeted drug delivery systems are drug carrier systems that deliver the drug to the target or receptor site in a manner that provides maximum therapeutic activity, prevents degradation or inactivation during transit to the target sites, and protects the body from adverse reactions because of inappropriate disposition.4,5 Design of an effective delivery system requires a thorough understanding of the drug, the disease, and the target site. Examples include macromolecular drug carriers (protein drug carriers), particulate drug delivery systems (eg, microspheres, nanospheres, and liposomes), monoclonal antibodies, and cells. Plasma clearance kinetics, tissue distribution, metabolism, and cellular interactions of a drug can be controlled by the use of a site-specific delivery system.6 Targeting of drugs to specific sites in the body can be achieved by linking particulate systems or macromolecular carriers to monoclonal antibodies or to cell specific ligands (eg, asialofetuin, glycoproteins, or immunoglobulins), or by alterations in the surface characteristics so that they are not recognized by the reticuloendothelial systems (RES).7 IMPORTANCE OF TARGETING Site-specific carriers guide the drug to the intended target site in which the receptors is located with exposing the drug to other tissues thereby avoiding adverse toxicity. The amount of drug needed will be reduced. APPROACHES TO DRUG TARGETING Basically there are four approaches Physical targeting: Involves the use of biologically active agents that are both potent & selective to a particular site in the body. Chemical targeting: Also known as prodrug approach. Preparation of pharmacologically inert forms of active drugs when they reach the active site they become activated by chemical or enzymatic reaction. Natural or passive targeting: Here a biologically inert micromolecular carrier system is utilized which direct the drug to a specific site where it accumulates and effects its response. Eg. Liposomes, nanoparticles etc… Active targeting: This is also known as ligand medicated targeting. Here the drug is attached to appropriate ligands. Eg. Monoclonal antibodies. RECENT APPROACHES Quantam dots A quantum dot is a semiconductor nanostructure that confines the motion of conduction band electrons, valence band holes, or excitons (bound pairs of conduction band electrons and valence band holes) in all three spatial directions. The confinement can be due to electrostatic potentials (generated by external electrodes, doping, strain, impurities), the presence of an interface between different semiconductor materials (e.g. in core-shell nanocrystal systems), the presence of the semiconductor surface (e.g. semiconductor nanocrystal), or a combination of these. Quantum dots are particularly significant for optical applications due to their theoretically high quantum yield. The ability to tune the size of quantum dots is advantageous for many applications and it is one of the most promising candidates for use in solid-state quantum computation and diagnosis , drug delivery, Tissue engineering, catalysis, filtration and also textiles technologies.7 Transdermal Approach Transdermal drug delivery system is topically administered medicaments in the form of patches that deliver drugs for systemic effects at a predetermined and controlled rate. A transdermal drug delivery device, which may be of an active or a passive design, is a device which provides an alternative route for administering medication. These devices allow for pharmaceuticals to be delivered across the skin barrier. In theory, transdermal patches work very simply. A drug is applied in a relatively high dosage to the inside of a patch, which is worn on the skin for an extended period of time. Through a diffusion process, the drug enters the bloodstream directly through the skin. Since there is high concentration on the patch and low concentration in the blood, the drug will keep diffusing into the blood for a long period of time, maintaining the constant concentration of drug in the blood flow.8 Folate Targeting Folate targeting is a method utilized in biotechnology for drug delivery purposes. It involves the attachment of the vitamin, folate (folic acid), to a molecule/drug to form a "folate conjugate". Based on the natural high affinity of folate for the folate receptor protein (FR), which is commonly expressed on the surface of many human cancers, folate-drug conjugates also bind tightly to the FR and trigger cellular uptake via endocytosis. Molecules as diverse as small radiodiagnostic imaging agents to large DNA plasmid formulations have successfully been delivered inside FR-positive cells and tissues. FA also displays high affinity for the folate receptor (FR), a glycosylphosphatidyinositol-linked protein that captures its ligands from the extracellular milieu and transports them inside the cell via a nondestructive, recycling endosomal pathway. The FR is also a recognized tumor antigen/biomarker. Because of this, diagnostic and therapeutic methods which exploit the FR’s function are being developed for cancer.8 Brain targeted drug delivery system The brain is a delicate organ, and evolution built very efficient ways to protect it. The delivery of drugs to central nervous system (CNS) is a challenge in the treatment of neurological disorders. Drugs may be administered directly into the CNS or systematically (e.g., by intravenous injection) for targeted action in the CNS. The major challenge to CNS drug delivery is the blood-brain barrier (BBB), which limits the access of drugs to the brain substance. Advances in understanding of the cell biology of the BBB have opened new avenues and possibilities for improved drug delivery to the CNS. Various strategies that have been used for manipulating the blood-brain barrier for drug delivery to the brain include osmotic and chemical opening of the blood-brain barrier as well as the use of transport/carrier systems. Other strategies for drug delivery to the brain involve bypassing the BBB. Various pharmacological agents have been used to open the BBB and direct invasive methods can introduce therapeutic agents into the brain substance. It is important to consider not only the net delivery of the agent to the CNS, but also the ability of the agent to access the relevant target site within the CNS. Various routes of administration as well as conjugations of drugs, e.g., with liposomes and nanoparticles, are considered.8 Liposomes These are vesicular concentric structures, range in size from a nanometer to several micrometers, containing a phospholipids bilayer and are biocompatible, biodegradable and non immunogenic. Liposomes have generated a great interest because of their versatility and have played a significant role in formulation of potent drugs to improve therapeutics. Enhanced safety and efficacy have been achieved for a wide range of drug classes, including antitumor agents, antiviral, antimicrobials, vaccines, gene therapeutics etc…Recently pharmaceutical science is using liposomes to reduce toxicity and side effect of drugs. The various problems like poor solubility, short half life and poor bioavailability & strong side effect of various drugs can be overcome by employing the concept of liposomes especially in various diseases like cancer etc. Liposomes offer ample opportunities for the investigators to explore the unidentified breakthrough in the field of pharmaceutical technology.9 Niosomes Niosomes are non-ionic surfactant vesicles that can entrap a solute in a manner analogous to liposomes.They are osmotically active, and are stable on their own, while also increasing the stability of the entrapped drugs.10,11 1. Handling and storage of surfactants require no special conditions. Niosomes possess an infrastructure consisting of hydrophilic and hydrophobic moieties together, and as a result, can accommodate drug molecules with a wide range of solubilities.12 2. Although niosomes as drug carriers have shown advantages such as being cheap and chemically stable, they are associated with problems related to physical stability such as fusion, aggregation, sedimentation and leakage on storage.13 3. All methods traditionally used for preparation of niosomes are time consuming and many involve specialized equipments. Most of these methods allow only for a predetermined lot size so material is often wasted if smaller quantities are required for particular dose application.14 4. The size of niosomes are microscopic and lies in nanometric scale. The particle size ranges from 10nm-100nm.15,16 Niosomes as drug carriers Several studies have described the properties of niosomes as drug carriers. Niosomes behave similarly to liposomes in vivo by prolonging circulation time of the encapsulated drug and altering chemical distribution within the body.17,18 However, niosomes have advantages over liposomes as drug carriers, including greater chemical stability, lower cost, easier storage and handling, and are less likely than liposomes to become toxic.19,20 Niosomal encapsulation reduces toxicity of drugs in many different applications and therapies. Niosomal drug delivery has been studied using various methods of administration,21 including intramuscular,22 intravenous23 peroral24 and transdermal25,26,27,28 Nebulized surfactants entrapping all-trans-retinoic acid (ATRA) were delivered as an inhaled aerosol reducing the drug toxicity and altering the pharmacokinetics. In addition, as drug delivery vesicles, niosomes have been shown to enhance absorption of some drugs across cell membranes to localize in targeted organs, tissues and to elude the reticulo endothelial system (RES).29 Cellular uptake of niosomes can be via endocytosis;12 however they have been shown to bind and fuse with cell plasma membranes via cellular receptors when vesicle surface charge is sufficiently negative.30 Method of Preparation of Niosomes Ether injection method In this method, a solution of the surfactant is made by dissolving it in diethyl ether. This solution is then introduced using an injection (14 gauge needle) into warm water or aqueous media containing the drug maintained at 60°C. Vaporization of the ether leads to the formation of single layered vesicles. The particle size of the Niosomes formed depend on the conditions used, and can range anywhere between 50-1000μm.31 Hand shaking method (Thin Film Hydration Technique) In this method a mixture of the vesicle forming agents such as the surfactant and cholesterol are dissolved in a volatile organic solvent such as diethyl ether or chloroform in a round bottom flask. The organic solvent is removed at room temperature using a rotary evaporator, which leaves a thin film of solid mixturedeposited on the walls of the flask. This dried surfactant film can then be rehydrated with the aqueous phase, with gentle agitation to yield multilamellar Niosomes. The multilamellar vesicles thus formed can further be processed to yield unilamellar Niosomes or smaller Niosomes using sonication, micro fluidization or membrane extrusion techniques.32 Reverse phase evaporation technique This method involves the creation of a solution of cholesterol and surfactant (1:1 ratio) in a mixture of ether and chloroform. An aqueous phase containing the drug to be loaded is added to this, and the resulting two phases are sonicated at 4-5°C. A clear gel is formed which is further sonicated after the addition of phosphate buffered saline (PBS). After this the temperature is raised to 40°C and pressure is reduced to remove the organic phase. This results in a viscous Niosome suspension which can be diluted with PBS and heated on a water bath at 60°C for 10 min to yield Niosomes.32 Trans membrane pH gradient (inside acidic) Drug Uptake Process (remote loading) In this method, a solution of surfactant and cholesterol is made in chloroform. The solvent is then evaporated under reduced pressure to get a thin film on the wall of the round bottom flask, similar to the hand shaking method. This film is then hydrated using citric acid solution (300mM, pH 4.0) by vortex mixing. The resulting multilamellar vesicles are then treated to three freeze thaw cycles and sonicated. To the niosomal suspension, aqueous solution containing 10mg/ml of drug is added and vortexed. The pH of the sample is then raised to 7.0-7.2 using 1M disodium phosphate (this causes the drug which is outside the vesicle to become non-ionic and can then cross the niosomal membrane, and once inside it is again ionized thus not allowing it to exit the vesicle). The mixture is later heated at 60°C for 10 minutes to give Niosomes.32 The Bubble Method It is a technique which has only recently been developed and which allows the preparation of Niosomes without the use of organic solvents. The bubbling unit consists of a round bottom flask with three necks, and this is positioned in a waterbath to control the temperature. Watercooled reflux and thermometer is positioned in the first and second neck, while the third neck is used to supply nitrogen. Cholesterol and surfactant are dispersed together in a buffer (pH 7.4) at 70°C. This dispersion is mixed for a period of 15 seconds with high shear homogenizer and immediately afterwards, it is bubbled at 70°C using the nitrogen gas to yield Niosomes.32 Formation of Proniosomes and Niosomes from Proniosomes To create Proniosomes, a water soluble carrier such as Sorbitol is first coated with the surfactant. The coating is done by preparing a solution of the surfactant with cholesterol in a volatile organic solvent, which is sprayed onto the powder of Sorbitol kept in a rotary evaporator. The evaporation of the organic solvent yields a thin coat on the Sorbitol particles. The resulting coating is a dry formulation in which a water soluble particle is coated with a thin film of dry surfactant. This preparation is termed Proniosome.33 Film Method The mixture of surfactant and cholesterol is dissolved in an organic solvent (e.g. diethylether, chloroform, etc.) in a round-bottomed flask. Subsequently, the organic solvent is removed by low pressure/vacuum at room temperature, for example using a rotary evaporator. The resultant dry surfactant film is hydrated by agitation at 50–60°C and multilamellar vesicles (MLV) are formed.33 Sonication Typically the aqueous phase is added into the mixture of surfactant and cholesterol in a scintillation vial. Then, it is homogenized using a sonic probe. The resultant vesicles are of small unilamellar (SUV) type Niosomes. The SUV type Niosomes are larger than SUV liposomes (i.e. SUV Niosomes are >100 nm in diameter while SUV liposomes are <100 nm in diameter). It is possible to obtain SUV Niosomes by sonication of MLV type vesicles, obtained for example through the film method explained above. For small volume samples probe type sonicator is used while for larger volume samples bath type sonicator is more appropriate.33 Method of Handjani–Vila Equivalent amounts of synthetic non-ionic lipids are mixed with the aqueous solution of the active substance to be encapsulated and a homogenous lamellar film is formed by shaking. The resultant mixture is homogenized employing ultracentrifugation and agitation at a controlled temperature. The Niosomes can be prepared from the Proniosome by adding the aqueous phase with the drug to the Proniosomes with brief agitation at a temperature greater than the mean transition phase temperature of the surfactant.33 Marketed Products Lancôme has come out with a variety of antiageing products which are based on noisome formulations. L’Oreal is also conducting research on anti-ageing cosmetic products. Niosomal Preparation in the Market is – Lancôme (www.lancome.com)34 Disadvantages of Niosomes 1. Physical instability 2. Aggregation 3. Fusion 4. Leaking of entrapped drug 5. Hydrolysis of encapsulated drugs which limiting the shelf life of the dispersion Proniosomes: To overcome all above Disadvantages of Niosomes, Proniosomes are prepared and reconstituted into Niosomes. Hu and Rhodes et al reported that Proniosomes are dry formulations of surfactant-coated carrier, which can be measured out as needed and rehydrated by brief agitation in hot water. These “proniosomes” minimize problems of niosomes physical stability such as aggregation, fusion and leaking and provided additional convenience in transportation, distribution, storage and dosing. Proniosome-derived niosomes are superior to conventional niosomes in convenience of storage, transport and dosing. Stability of dry proniosomes is expected to be more stable than a pre-manufactured niosomal formulation. In release studies proniosomes appear to be equivalent to conventional niosomes. Size distributions of proniosome-derived niosomes are somewhat better that those of conventional niosomes so the release performance in more critical cases turns out to be superior.35 Proniosomes are dry powder, which makes further processing and packaging possible. The powder form provides optimal flexibility, unit dosing, in which the proniosome powder is provided in capsule could be beneficial. A proniosome formulation based on maltodextrin was recently developed that has potential applications in deliver of hydrophobic or amphiphilic drugs. The better of these formulations used a hollow particle with exceptionally high surface area. The principal advantage with this formulation was the amount of carrier required to support the surfactant could be easily adjusted and proniosomes with very high mass ratios of surfactant to carrier could be prepared. Because of the by slurry method, hydration of surfactant from proniosomes of a wide range of compositions can be studied.36 Advantages of proniosomes over the niosomes Avoiding problem of physical stability like aggregation, fusion, leaking. Avoiding hydrolysis of encapsulated drugs which limiting the shelf life of the dispersion. Commonly used materials for proniosomes preparation36 Surfactants: Span20, Span40, Span60, Span80, Span85, Tween20, Tween40, Tween80 Stabilizers: Cholesterol, lecithin Carriers: Maltodextrin, sorbitol, mannitol, magnesium aluminum silicate, microcrystalline cellulose, spray dried lactose, glucose monohydrate and sucrose Stearates Selection of the carrier in the proniosomal formulation requires more attention as it affects some factors like flexibility in surfactant and other component ratio, surface area, efficient loading, etc… Method of Preparation of proniosomes The proniosomes can be prepared by 1. Spraying method. 2. Slurry method. Spraying method (Hu and Rhodes et al in 1999) prepared proniosomes by spraying the surfactant in organic solvent onto sorbitol powder and then evaporating the solvent. Because the sorbitol carrier is soluble in the organic solvent, it is necessary to repeat the process until the desired surfactant load has been achieved. The surfactant coating on the carrier comes out to be very thin and hydration of this coating allows multilamellar vesicles to form.37 Slurry method (A lmir.I and Blazek - Walsh et al in 2001) developed slurry method to produce proniosomes using maltodextrin as a carrier. The time required to produce proniosome by this is independent of the ration of surfactant solution to carrier material. In slurry method, the entire volume of surfactant solution is added to maltodextrin powder in a rotary evaporator and vacuum applied until the powder appears to be dry and free flowing. Drug containing proniosome-derived niosomes can be prepared in manner analogous to that used for the conventional niosomes, by adding drug to the surfactant mixture prior to spraying the solution onto the carrier (sorbitol, maltodextrin) or by addition of drug to the aqueous solution used to dissolve hydrate the proniosomes.37 Formation of Niosomes from Proniosomes The niosomes can be prepared from the proniosomes by adding the aqueous phase with the drug to the proniosomes with brief agitation at a temperature greater than the mean transition phase temperature of the surfactant. Fig:1 Formation of Niosomes from Proniosomes. T > Tm Where, T = Temperature Tm = mean phase transition temp Blazek-Walsh A.I. et al has reported the formulation of niosomes from maltodextrin based proniosomes. This provides rapid reconstitution of niosomes with minimal residual carrier. Slurry of maltodextrin and surfactant was dried to form a free flowing powder, which could be rehydrated by addition of warm water.37,38 Characterization of proniosomes Measurement of Angle of repose The angle of repose of dry proniosomes powder was measured by a funnel method (Lieberman et al 1990). The proniosomes powder was poured into a funnel which was fixed at a position so that the 13mm outlet orifice of the funnel is 5cm above a level black surface. The powder flows down from the funnel to form a cone on the surface and the angle of repose was then calculated by measuring the height of the cone and the diameter of its base.39 Scanning electron microscopy Particle size of proniosomes is very important characteristic. The surface morphology (roundness, smoothness, and formation of aggregates) and the size distribution of proniosomes were studied by Scanning Electron Microscopy (SEM).Proniosomes were sprinkled on to the double- sided tape that was affixed on aluminum stubs. The aluminum stub was placed in the vacuum chamber of a scanning electron microscope (XL 30 ESEM with EDAX, Philips, Netherlands). The samples were observed for morphological characterization using a gaseous secondary electron detector (working pressure: 0.8 torr, acceleration voltage: 30.00 KV) XL 30, (Philips, Netherlands).39 Optical Microscopy The niosomes were mounted on glass slides and viewed under a microscope (Medilux207RII, Kyowa-Getner, Ambala, India) with a magnification of 1200X for morphological observation after suitable dilution. The photomicrograph of the preparation also obtained from the microscope by using a digital SLR camera.39 Measurement of vesicle size The vesicle dispersions were diluted about 100 times in the same medium used for their preparation. Vesicle size was measured on a particle size analyzer (Laser diffraction particle size analyzer, Sympatec, Germany). The apparatus consists of a He-Ne laser beam of 632.8 nm focused with a minimum power of 5mW using a Fourier lens [R-5] to a point at the center of multielement detector and a small volume sample holding cell (Su cell). The sample was stirred using a stirrer before determining the vesicle size. Hu C. and Rhodes in 1999 reported that the average particle size of proniosomes derived niosomes is approximately 6μm while that of conventional niosomes is about 14μm.39,40 Entrapment efficiency Entrapment efficiency of the niosomal dispersion in can be done by separating the unentrapped drug by dialysis, centrifugation or gel filtration as described above and the drug remained entrapped in niosomes is determined by complete vesicle disruption using 50% npropanol or 0.1% Triton X-100 and analyzing the resultant solution by appropriate assay method for the drug.39,40 In vitro drug release can be done by (Chen DB et al., 2001) Dialysis tubing Reverse dialysis Franz diffusion cell Dialysis tubing Muller et al in 2002 studied in vitro drug release could be achieved by using dialysis tubing. The proniosomes is placed in prewashed dialysis tubing which can be hermetically sealed. The dialysis sac is then dialyzed against a suitable dissolution medium at room temperature; the samples are withdrawn from the medium at suitable intervals, centrifuged and analysed for drug content using suitable method (U.V. spectroscopy, HPLC etc). The maintenance of sink condition is essential.41,42 Reverse dialysis In this technique a number of small dialysis as containing 1ml of dissolution medium are placed in proniosomes. The proniosomes are then displaced into the dissolution medium. The direct dilution of the proniosomes is possible with this method; however the rapid release cannot be quantified using this method.42 Franz diffusion cell The in vitro diffusion studies can be performed by using Franz diffusion cell. Proniosomes is placed in the donor chamber of a Franz diffusion cell fitted with a cellophane membrane. The proniosomes is then dialyzed against a suitable dissolution medium at room temperature; the samples are withdrawn from the medium at suitable intervals, and analysed for drug content using suitable method (U.V spectroscopy, HPLC, etc) .the maintenance of sink condition is essential.41,43 Drug Release Kinetic Data Analysis The release data obtained from various formulations were studied further for their fitness of data in different kinetic models like Zero order, Higuchi’s and peppa’s. In order to understand the kinetic and mechanism of drug release, the result of in-vitro drug release study of Niosome were fitted with various kinetic equation like zero order (Equation 1) as cumulative % release vs. time, higuchi’s model (Equation 2) as cumulative % drug release vs. square root of time. r2 and k values were calculated for the linear curve obtained by regression analysis of the above plots C = k0t …..(1) Where k0 is the zero order rate constant expressed in units of concentration / time and t is time in hours. Q = kHt1/2 …..(2) Where kH is higuchi’s square root of time kinetic drug release constant. To understand the release mechanism in-vitro data was analyzed by peppa’s model (Equation 3) as log cumulative % drug release vs. log time and the exponent n was calculated through the slope of the straight line. Mt / M∞ = btn …..(3) Where Mt is amount of drug release at time t, M∞ is the overall amount of the drug, b is constant, and n is the release exponent indicative of the drug release mechanism. If the exponent n = 0.5 or near, then the drug release mechanism is Fickian diffusion, and if n have value near 1.0 then it is non-Fickian diffusion.44 Osmotic shock The change in the vesicle size can be determined by osmotic studies. Niosomal formulations are incubated with hypotonic, isotonic, hypertonic solutions for 3 hours. Then the changes in the size of vesicles in the formulations are viewed under optical microscopy.45 Stability studies To determine the stability of proniosomes, the optimized batch was stored in airtight sealed vials at different temperatures. Surface characteristics and percentage drug retained in proniosomes and proniosomes derived niosomes were selected as parameters for evaluation of the stability, since instability of the formulation would reflect in drug leakage and a decrease. in the percentage drug retained.45 the proniosomes were sample at regular intervals of time (0,1,2,and 3months ),observed for color change, surface characteristics and tested for the percentage drug retained after being hydrated to form niosomes and analysed by suitable analytical methods(UV spectroscopy, HPLC methods etc).46 Zeta potential analysis Zeta potential analysis is done for determining the colloidal properties of the prepared formulations. The suitably diluted proniosome derived noisome dispersion was determined using zeta potential analyzer based on electrophoretic light scattering and laser Doppler velocimetry method (Zetaplus™, Brookhaven Instrument Corporation, New York, USA).46 The temperature was set at 25°C. Charge on vesicles and their mean zeta potential values with standard deviation of 5 measurements were obtained directly from the measurement.47 Applications of Proniosomes: Drug Targeting: One of the most useful aspects of proniosomes is their ability to target drugs. Proniosomes can be used to target drugs to the reticulo-endothelial system. The reticulo-endothelial system (RES) preferentially takes up proniosome vesicles. The uptake of proniosomes is controlled by circulating serum factors called opsonins. These opsonins mark the proniosome for clearance. Such localization of drugs is utilized to treat tumors in animals known to metastasize to the liver and spleen. This localization of drugs can also be used for treating parasitic infections of the liver. Proiosomes can also be utilized for targeting drugs to organs other than the RES. A carrier system (such as antibodies) can be attached to proniosomes (as immunoglobulin bind readily to the lipid surface of the niosome) to target them to specific organs.47 Many cells also possess the intrinsic ability recognize and bind specific carbohydrate determinants, and this can be exploited by proniosomes to direct carrier system to particular cells.48,49 1. Anti-neoplastic Treatment: Most antineoplastic drugs cause severe side effects. Niosomes can alter the metabolism; prolong circulation and half life of the drug, thus decreasing the side effects of the drugs. Niosomal entrapment of Doxorubicin and Methotrexate50,51,52 (in two separate studies) showed beneficial effects over the unentrapped drugs, such as decreased rate of proliferation of the tumor and higher plasma levels accompanied by slower elimination.49 Podophyllotoxin-dipalmitoylphosphatidylcholine (PPT-DPPC) proliposomes (PPT-DPPCPL) for improvement of the stability of PPT-DPPC liposome. Methods Freeze-drying method was used to prepare PPT-DPPC-PL, and the particle morphology, size range, encapsulation efficiency and stability of PPT-DPPC lip osome were investigated. Results After hydration of PPTDPPC-PL, PPT-DPPC liposome appeared multivesicular under electron microscope and the particles were distributed homogenously with an average particle size of 1.45±0.38 μm. The encapsulation efficiency of PPT was 72.3%, and alters storage at 4 to 40 for 4 to 6 months, the proliposome remained stable. Conclusion the prepared PPT-DPPC-PL particles by freeze-drying method are evenly distributed. The preparation method is relatively simple with higher embedding ratio and better stability.53 2. Leishmaniasis: Leishmaniasis is a disease in which a parasite of the genus Leishmania invades the cells of the liver and spleen. Commonly prescribed drugs for the treatment are derivatives of antimony (antimonials), which in higher concentrations can cause cardiac, liver and kidney damage. Use of pronoisome in tests conducted showed that it was possible to administer higher levels of the drug without the triggering of the side effects, and thus allowed greater efficacy in treatment.54,55 3. Delivery of Peptide Drugs: Oral peptide drug delivery has long been faced with a challenge of bypassing the enzymes which would breakdown the peptide. Use of niosomes to successfully protect the peptides from gastrointestinal peptide breakdown is being investigated.55 In an invitro study conducted by Yoshida et al, oral delivery of a vasopressin derivative entrapped in niosomes showed that entrapment of the drug significantly increased the stability of the peptide.56 4. Uses in Studying Immune Response: Proniosomes are used in studying immune response due to their immunological selectivity, low toxicity and greater stability. Niosomes are being used to study the nature of the immune response provoked by antigens.56 5. Proniosomes as Carriers for Haemoglobin: (Moser P. and Marchand Arvier M. in 1989) reported that niosomes can be used as carriers for haemoglobin within the blood.56 The niosomal vesicle is permeable to oxygen and hence can act as a carrier for hemoglobin in anemic patients.57 6. Proniosomes used in Cardiac Disorders: Proniosomal carrier system for captopril for the treatment of hypertension that is capable of efficiently delivering entrapped drug over an extended period of time. The potential of proniosomes as a transdermal drug delivery system for captopril was nvestigated by encapsulating the drug in various formulations of proniosomal gel composed of various ratios of sorbitan fatty acid esters, cholesterol, lecithin prepared by coacervation-phase separation method.57,58 7. Sustained Release: Azmin et al suggested the role of liver as a depot for methotrexate after niosomes are taken up by the liver cells.59 Sustained release action of niosomes can be applied to drugs with therapeutic index and low water solubility since those could be maintained in the circulation via niosomal encapsulation.60 8. Localized Drug Action: Drug delivery through niosomes is one of the approaches to achieve localized drug action, since their size and low penetrability through epithelium and connective tissue keeps the drug localized at the site of administration.61 Localized drug action results in enhancement of efficacy of potency of the drug and at the same time reduces its systemic toxic effects e.g. Antimonials encapsulated within niosome are taken up by mononuclear cells resulting in localization of drug, increase in potency and hence decrease both in dose and toxicity. The evolution of niosomal drug delivery technology is still at an infancy stage, but this type of drug delivery system has shown promise in cancer chemotherapy and antileishmanial therapy.61,62 9. Antibacterial treapy: Amphotericin-b proliposomes could be stored for 9 months without significant changes in distribution of vesicle size and for 6 months without loss of pharmacological activity. Even though physical stability of the preparation can be increased, a vacuum or nitrogen atmosphere is still required during preparation and storage to prevent oxidation of phospholipid.62,63 10. Hormonal Therapy: A proniosome based transdermal drug delivery system of levonorgestrel (LN) was developed and extensively characterized both in vitro and in vivo. The proniosomal structure was liquid crystallinecompact niosomes hybrid which could be converted into niosomes upon hydration. The system was evaluated in vitro for drug loading, rate of hydration (spontaneity), vesicle size, polydispersity, entrapment efficiency and drug diffusion across rat skin. The effect of composition of formulation, amount of drug, type of Spans, alcohols and sonication time on transdermal permeation profile was observed. The stability studies were performed at 4°C and at room temperature.61,62 The biological assay for progestational activity included endometrial assay and inhibition with the formation of corpora lutea. The study demonstrated the utility of proniosomal transdermal patch bearing levonorgestrel for effective contraception.63 11. Cosmetics/Cosmeceuticals: Proniosomal gels are generally present in transparent, translucent or white semisolid gel texture, which makes them physically stable during storage and transport. Due to the limited solvent system present, the proniosomes formed were the mixture of many phases of liquid crystal, viz. lamellar, hexagonal and cubic phase liquid crystals. Dissolution of most surfactants in water, leads to the formation of lyotropic liquid crystals rather than micellar solution. Lamellar phase shows sheets of surfactants arranged in bilayer form, whereas in hexagonal phase cylindrical units are packed in hexagonal fashion. Cubic phase consists of curved bio-continuous lipid bilayer extending in three dimensions, separating two congruent networks of water channels. These liquid crystals present an attractive appearance because of their, transparency and high viscosity, although in the beginning of its formation, a short range of less viscous compositions (so called liquid/gel compositions) appear in some cases. Addition of water leads to interaction between water and polar groups of the surfactant results in swelling of bilayer. Proniosomes exists in two forms, i.e. semisolid liquid crystal gel and dry granular powder, depending on their method of preparation.64,65,66 12. NSAID appllication: Ketorolac tromethamine (KT) is a nonsteroidal agent with potent analgesic and moderate anti-inflammatory activity. The drug is currently administered intramuscularly and orally in divided multiple doses for short-term management of postoperative pain (30 mg q.i.d. by IM injection and 10 mg q.i.d. as oral tablets).66 This frequent dosing, which results in unacceptable patient compliance, is required due to the short half-life of the drug (4–6 h). Therefore, an alternative noninvasive mode of delivery of the drug is needed. Transdermal delivery certainly appears to be an attractive route of administration to maintain the drug blood levels of KT for an extended period of time.67 13. As Antiallergic: Investigate the preparation and safety of carboplatin proliposomes Methods: Carboplatin liposomes were prepared by film extrusion. Mannitol was incorporated into the liposome to prevent the liposome vesicles from aggregation. The carboplatin proliposomes were obtained by lyophilization.Allergic reactions to carboplatin proliposomes were evaluated in Guinea pigs. The vein irritation and hemolysis post the administration of liposomes in rabbits were characterized.68 14. Preparation of Vitamins: To prepare vitamin A proliposomes and to enhance stability of vitamin A. Methods: Freeze drying method was used to prepare vitamin A proliposomes. The particle morphology, the size range, encapsulation efficiency and stability of vitamin A liposomes were studied.69 15. Co-Enzyme preparation of Proliposome: To enhance the stability of coenzyme Q10 liposomes, freeze-drying method was used to prepare coenzyme Q10 proliposomes. Sucrose, trehalose, mannitol and lactose were selected as cryoprotectant. The particle morphology, the size range and encapsulation efficiency of coenzyme Q10 liposomes before freeze-drying and after hydrated were studied. The weight ratios of the optimized formulation were trehalose egg lecithin 4 to1. After coenzyme Q10 proliposomes was hydrated, the average diameter of rehydration liposomes was 508nm,and the encapsulation efficiency was about 75%.The analysis of FT-IR spectrum showed that the hydrogen bond between trehalose and lipid head groups formed in the dry state.70 REFERENCES 1. Chien Y.W., Novel drug delivery systems, Drugs and the Pharmaceutical Sciences, 50, New York, 797, 992 (2008) 2. Allen T. M. and Cullis P. R., Drug Delivery Systems, Entering the Mainstream Science, 1818-1822 (2004) 3. Nacht S. and Kantz M, A., Novel Topical Programmable Delivery System, Topical Drug Delivery Systems, 299-325 (1992) 4. Won R., Method for delivering an active ingredient by controlled time release utilizing a novel delivery vehicle, 825 (1987) 5.Gregoriadis, G.,Targeting of drug, Nature (1977)265 407–411. 6. Gregoriadis, G., Florence, A.T., Clinical diagnostic and ophthalmic potential, Drugs (1993) 45, 15–28. 7. Pozanansky,M.J., Juliano,R.,L., Pharmacol. Rev. 36 (1984) 277–336. 8. Roland Bodmeier., 1998. Polymeric Dispersion as Drug Carriers, In: Pharmaceutical Dosage Forms: Disperse System, Vol-3 (A. Liberman, ed.), Marcel Dekker, New York, 87127. 9. Sung-Ho Kim, 2002. Development of a polymeric nanoparticulate drug delivery system In vitro characterization of nanoparticles based on sugar containing conjugates. Int. J. Pharm. 245, 67-73. 10. Chain, Y. W., 1992. Oral Drug Delivery and Delivery System. In, Novel Drug Delivery System, 2nd Edn (James Swarbrick, ed.), Marcel Dekker, New York, 139-196. 11. Brahmankar, D. M., and Jaiswal, S. B., 1995. In; Biopharmaceutics and Pharmacokinetics A Treatise, 1st Edn., Vallabh Prakashan Publications, Delhi, 1995,50-57. 12. Baillie AJ, Florence AT, Hume LR, Muirhead GT and Rogerson A. The preparation and properties of niosomes non-ionic surfactant vesicles. J. Pharm. Pharmacol. (1985) 37: 863868 13. Rogerson A, Cummings J, Willmott N and Florence AT. The distribution of doxorubicin in mice following administration in niosomes. J. Pharm. Pharmacol. (1988) 40: 337-342 14. Biju SS, Talegaonkar S, MisraPR and Khar RK. Vesicular systems: An overview. Indian J. Pharm. Sci. (2006) 68: 141-153 15. Ijeoma, F., Uchegbu, Suresh P.Vyas., 1998. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 172, 33–70. 16. Malhotra M. and Jain N.K. Niosomes as Drug Carriers. Indian Drugs (1994), 31 (3): 8186 17. Azmin M.N., Florence A.T., Handjani-Vila R.M., Stuart J.F.B., Vanlerberghe G., and Whittaker J.S. The effect of non-ionic surfactant vesicle (niosome) entrapment on the absorption and distribution of methotrexate in mice. J. Pharm. Pharmacol. 1985; 37: 237– 242. 18. Ruckmani, K., Jayakar, B., Ghosal, S.K., 2002. Nonionic surfactant vesicles (niosomes) of cytarabine hydrochloride for effective treatment of leukemias: encapsulation, storage, and in vitrorelease.DrugDev.Ind.Pharm.26, 217–222. 19. Uchegbu, I.F., Double, J.A., Turton, J.A., Florence, A.T., 1995. Niosome encapsulation of a doxorubicin polymer conjugates. Pharmaceutical Research 12, 1019–24. 20. Almira, I., Blazek-Welsh., Rhodes, D. G., 2001. Maltodextrin-Based Proniosomes. AAPS PharmSciTech 3 (1) article 1. 21. Blazek-Walsh A.I. and Rhodes D.G. Pharm. Res. SEM imaging predicts quality of niosomes from maltodextrin-based proniosomes. 2001; 18: 656-661. 22. Runothayanun, P., Sooksawate, T., Florence, A.T., 1999. Extrusion of niosomes from capillaries: approaches to pulsed delivery device. J. Control. Release. 60 (2), 391-397. 23. Namdeo, A., Jain, N.K., 1999. Niosomal delivery of 5-fluorouracil. J. Microencapsulation 16, (6), 731 – 740. 24. Rentel, C.O., Bouwstra, J.A., Naisbett, B., Junginger, H.E., 1999. Niosomes as a novel peroral vaccine delivery system. Int. J. Pharm. 186, 161–167. 25. Yoshioka T., Stermberg B. and Florence A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60, and 80) and a sorbitan triester (Span 85). Int J Pharm. 1994; 105:1-6. 26. Ajay Solanki, Jolly Parikh and Rajesh Parikh. Preparation, Characterization, optimization, and stability studies of Aceclofenac Proniosomes Iranian J. Pharm Research 2008; 7(4):237246. 27. Mahdi, Jufri, Effionora, Anwar, Joshita, Djajadisastra, 2004. Preparation of Maltodextrin DE 5-10 based ibuprofen Proniosomes. Majalah Ilmu Kefarmasian I, 1, 10 – 20 28. Solanki, A. B., Parikh, J. R., Parikh, R.H., 2007. Formulation and Optimization of Piroxicam Proniosomes by 3-Factor, 3-Level Box-Behnken Design. AAPS PharmSciTech. 8(4), 86, E1-E7. 29. Goopi N. Devaraj., Prakash, S. R., Ravi Devaraj, Apte, S.S., B. Ramesh Rao., D.Rambhav. 2002. Release studies on Niosomes containing fatty alcohols as bilayer stabilizers instead of cholesterol. Journal of colloids and interface science. 251, 360-365. 30. Elsie Oommen., Sandip, B., Tiwari, Udupa, N., Ravindra Kamath., Uma Devi, P., 1999. Niosome entrapped β-cyclodextrin methotrexate complex as a drug delivery system. Indian J. Pharmacol. 31, 279-284 31. J.Grait, R.M.handjani A.Ribier, G.Vanlerberghe, A.Zabotto and pharmaceutical compositions containing niosomes and a water soluble polyamide and a process for preparing these compositions. US patent No 4830857, L’OREAL, US patent no 4830857. 32. Suggy s, murari CR, Ahmad. Niosomes (a review) Biopharm, 2004, 24-37 33. Lasic DD, Niosomes: From physics to Application. Amsterdam: Elsevier, 1993. 34. Ajay Solanki, Jolly Parikh and Rajesh Parikh. Preparation, Characterization, optimization, and stability studies of Aceclofenac Proniosomes Iranian J. Pharm Research 2008; 7(4):237246. 35. A. Alsarra, A. A. Bosela, S.M. Ahmed, and G. M. Mahrous. Proniosomes as a drug carrier for transdermal delivery of ketorolac. Eur. J. Pharm. and Biopharm. Xx: 1–6(2004) 36. Hu C. and Rhodes D.G. Proniosomes: a novel drug carrier preparation. Int. J. Pharm. 1999; 185: 23-35. 37. Chauhan S. and Luorence M.J. The preparation of polyoxyethylene containing non-ionic surfactant. Vesicles. J. Pharm. Pharmacol. 1989; 41: 6p. 38. Gayatri Devi S., Venkatesh P. and Udupa N. Niosomal sumatriptan succinate for nasal administration. Int. J. Pharm. Sci. 2000; 62(6), 479-481. 39. Chandraprakash K.S., Udupa N., Umadevi P. and Pillai G.K. Pharmacokinetic evaluation of surfactant vesicles containing methotrexate in tumor bearing mice. Int. J. Pharma. 1990; R1- R3: 61. 40. B.Vora, A.J. Khopade, N.K. Jain, Proniosome based transdermal delivery of levonorgestrel for effective contraception, J. Control.Release 54 (1998) 149–165. 41. Puglia C, Trombetta D, Venuti V, Saija A, Bonina F (2004). Evaluation of in vivo topical anti-inflammatory activity of indometacin from liposomal vesicles. J. Pharm. Pharmacol. 56: 1225-1232. 42. R.H.Muller, M. Radtke, S.A. Wissing, Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations, Adv. Drug Deliv. Rev. 54 (2002)131–155. 43. Vyas S.P., Khar R,K., Niosomes Targeted and Controlled Drug delivery,249 – 279. 44. Gibaldi .M and Perrier D; Pharmacokinetics second edition, New York, Marcel Dekker, Inc., 1982. 45. Biju SS, Talegaonkar S, MisraPR and Khar RK. Vesicular systems: An overview. Indian J. Pharm. Sci. (2006) 68: 141-153 46. Effect of Charged and Non-ionic Membrane Additives on Physicochemical Properties and Stability of Niosomes Varaporn Buraphacheep Junyaprasert,Veerawat Teeranachaideekul, and Tasaneeya Supaperm1 AAPS PharmSciTech, Vol. 9, No. 3, September 2008. 47. Gregoriadis G. Targeting of drugs: implications in medicine. Lancet. 1981; 2(8240): 241246. 48. Solanki A B, Parikh J R, Parikh RH, 2007. Formulation and Optimization of Piroxicam Proniosomes by 3-Factor, 3-Level Box- Behnken Design. AAPS PharmSciTech. 8(4),86, E1E7. 49. Sudhamani.T, Priyadarisini.N, Radhakrishnan, Proniosomes – A Promising Drug Carriers, International Journal of PharmTech Research CODEN (USA): IJPRIF ISSN : 09744304 Vol.2, No.2, pp 1446-1454, April-June 2010; 50. Elsie Oommen., Sandip, B., Tiwari, Udupa, N., Ravindra Kamath., Uma Devi, P., 1999. Niosome entrapped β-cyclodextrin methotrexate complex as a drug delivery system. Indian J. Pharmacol. 31, 279-284 51. Chandrprakash K.S., Udupa N., Umadevi P. and Pillai G.K. Pharmacokinetic evaluation of surfactant vesicles containing methotrexate in tumor bearing mice. Int. J. Pharma. 1990; R1- R3: 61. 52. Varaporn BJ, Veerawat T, andTasaneeya S, Effect of Charged and Non-ionic Membrane Additives onPhysicochemical Properties andStability of Niosomes , 1AAPS PharmSciTech, 2008; September Vol. 9, No. 3 53. LIN Zhong-fang~1, ZENG Kang~1, ZHOU Zai-gao~1, LI Guo-feng~2, XIE Fangming~1, ZHU Xiao-liang~1, ZHANG San-quan~1 Department of Dermatology~1, Department of Pharmacy~2, Nanfang Hospital, First Military Medical University, Guangzhou 510515, China;Preparation and characterization of Podophyllotoxindipalmitoylphosphatidylcholine proliposome[J];Journal of First Military Medical University;2004-07. 54. Swati G, Ajay P, etal ,Drug Delivery Strategies for visceral Leishmaniasis, Expert Opin Drug Deliy, 2010; Mar.7 (3):371-402 20201740. 55. Hunter CA, Dolan TF, Coombs GH and Baillie AJ. Vesicular systems (niosomes andliposomes) for delivery of sodium stibogluconate in experimental murine visceral leishmaniasis.J.Pharm. Pharmacol. 1988; 40(3): 161-165. 56. Chain YW, 1992. Oral Drug Delivery and Delivery System. In, Novel Drug Delivery System, 2nd Edn (James Swarbrick, ed.), Marcel Dekker, New York, 139-196. 57. Moser P, Marchand-Arvier M, Labrude P, Handjani Vila. R.M. and Vignerson C.Niosomes d' hémoglobine. I. Preparation,proprietes physicochimiques et oxyphoriques,stabilite. Pharma. Acta.Helv. 1989; 64 (7): 192- 202. 58. Gupta et al, Transdermal Drug Delivery System for Captopril , 687 Trop J Pharm Res, June 2007; 6 (2) 59. Thakur R, Anwer MK, Shams MS Ali A, Khar RK, Shakeel F, Taha EI. Proniosomal transdermal therapeutic system of losartan potassium: development and pharmacokinetic evaluation. J Drug Target. 2009 Jul; 17(6):442-449. 60. Almira I, Blazek-Welsh, Rhodes DG, 2001. Maltodextrin-Based Proniosomes. AAPS PharmSciTech 3 (1) article 1. 61. Goopi N. Devaraj, Prakash, SR, Ravi Devaraj, Apte SS, B Ramesh Rao, D Rambhav. 2002. Release studies on Niosomes containing fatty alcohols as bilayer stabilizers instead of cholesterol. Journal of colloids and interface science. 251, 360-365 62. Jain, NK, Suman, Ramteke RB, UmaMaheshwari, 2006. Clarithromycin based oralsustained release nanoparticulate drug delivery stystem. Indian J. Pharm. Sci. 68 (4), 479 63. Elsie Oommen, Sandip B, Tiwari, Udupa N, Ravindra Kamath, Uma Devi, P, 1999.Niosome entrapped β-cyclodextrin methotrexate complex as a drug delivery system. Indian J.Pharmacol. 31, 279-284 19. Chandraprakash K.S. 64. Kumar S, etal, Site directed drug delivery by non-viral mode, Indian Journal of Biotechnology Vol. 6, April 2007; pp 159-174. 65. Blazek-Walsh AI and Rhodes DG. Pharm. Res. SEM imaging predicts quality of niosomes from maltodextrin-based proniosomes. 2001;18: 656-661. 66. Souto EB, Muller RH. Cosmetic features and application of lipid nanoparticles (SLN, NLC). Int J Cosmetic Science 2008; 30, 157-165. 67. Mahdi, Jufri, Effionora, Anwar, Joshita, Djajadisastra, 2004. Preparation of Maltodextrin DE 5-10 based ibuprofen Proniosomes. Majalah Ilmu Kefarmasian I, 1, 10 – 20 68. TIAN Wei-wei LIU Qing-fei LUO Guo-an WANG Yi-ming SU Qiang ,Preparation and Preliminary safety evaluation of carboplatin Proliposome , Chinese Journal of New Drugs 2006-13. 69. CHEN Jian Ming *, ZHANG Yang Mei, GAO Shen, ZHONG Yan Qiang, ZHANG Ya Ming (Department of Pharmaceutics, School of Pharmacy, Second Military Medical University, Shanghai 200433,China);Preparation of vitamin A proliposomes by freeze-drying method[J];Academic Journal of Second Military Medical University;2003-02. 70. Xia Shuqin et al;Preparation of coenzyme Q_(10) nanoliposomes[J];Science and Technology of Food Industry;2006-02 MARKETED PRODUCTS Name of therapeutic agent Therapeutic category Route of delivery Delivery system Captopril Antihypertensive Transdermal Proniosomal gel Flurbiprofen NSAIDs Transdermal Proniosomal gel Furesemide Antihypertensive Transdermal Proniosomal gel Lasartan Antihypertensive Transdermal Proniosomal gel