Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Fabry disease (angiokeratoma corporis
diffusum universale)
Johannes Fabry
Anderson-Fabry disease, also named Fabry disease (DF) or angiokeratoma corporis
diffusum universale (OMIM #301500), was reported independently and almost
simultaneously by dermatologists William Anderson in England and Johannes Fabry in
Germany, in 1898 1-3.
Fabry disease
•
Fabry disease (also known as Fabry's disease, Anderson-Fabry
disease, angiokeratoma corporis diffusum, and alpha-galactosidase A
deficiency) is a rare genetic lysosomal storage disease, inherited in an Xlinked manner. Fabry disease can cause a wide range of systemic
symptoms. It is a form of sphingolipidosis, as it involves dysfunctional
metabolism of sphingolipids. The disease is named after one of its
discoverers, Johannes Fabry (June 1, 1860 – June 29, 1930).
•
•
•
•
•
•
•
Synonyms of Fabry Disease
alpha-galactosidase A deficiency
Anderson-Fabry disease
angiokeratoma corporis diffusum
angiokeratoma diffuse
ceramide trihexosidase deficiency
GLA deficiency
Fabry disease
CLINICAL PRESENTATION
•
Clinical signs and symptoms of FD are very heterogeneous and subtle at first, which hinders or
delays the diagnosis in many situations.
•
• Homozygote: in general, there is total loss of enzyme function and they have the classical form
of the disease. At first, symptoms occur in childhood and adolescence, including chronic
paresthesia and episodes of acral and/or abdominal pain (Fabry crisis), heat intolerance,
reduction or absence of sudoresis, presence of angiokeratomas (AK) on the skin and/or mucosa
and cornea verticilata. Between the third and fourth decades of life, there is increase in previously
mentioned symptoms and there are also reports of progressive systemic impairment (heart, renal
and brain abnormalities) . In the absence of family history, diagnosis takes long time to be made
(mean age of 29 years), when there is irreversible visceral damage . Milder forms of the disease,
which are manifested later with primary affection of renal or cardiovascular systems, are known as
renal or heart variant, respectively, or atypical forms of FD and occur in patients with detectable
enzymatic activity . Forms of intermediate severity, between the classical phenotype and renal or
heart variants were described and are named intermediate forms.
•
• Heterozygote: clinical presentation of the disease in women is variable and it may range from
asymptomatic status to as severe presentations as those in men .
Patients that have blood antigen B have clinical manifestations that are relatively more severe,
given that degradation of this antigen is also dependent on enzyme α-GAL.
•
•
DERMATOLOGICAL FINDINGS
•
•
•
•
•
•
•
•
Angiokeratomas (tiny, painless papules that can appear on any region of the body, but are predominant on the
thighs, around the belly button, buttocks, lower abdomen, and groin) are common. Clinically, they are presented as
innumerous papules of varied color, from red to black, with slightly keratosic surface and variable size between 1
to 10 mm in diameter. They tend to be arranged in groups and normally they have symmetrical distribution
preferably on the skin area that goes from the navel to the tights (distribution named "bathing suit") The number
and size of AK increases as time goes by,, and they are not always directly related with systemic morbidity. They
are persistent lesions, but some AK may progress to thrombosis and disappear spontaneously.
Anhidrosis (lack of sweating) is a common symptom, and less commonly hyperhidrosis . AK are vascular lesions
that comprehend one or more dilated blood vessels on the upper dermis, directly under the epidermis, which are
normally followed by epidermal reactions characterized by acanthosis and/or hyperkeratosis
Additionally, patients can exhibit Raynaud's disease-like symptoms with neuropathy (in particular, burning
extremity pain).
Mucosa can also be affected in FD. As a result of disease advance, we can commonly see AK in the mucosa,
especially in the oral mucosa, affecting the anterior portion of gengival mucosa or labial mucosa, displaying a
telangiectasic aspect
Ocular involvement may be present showing cornea verticillata (also known as vortex keratopathy), i.e. clouding of
the corneas. Keratopathy may be the presenting feature in asymptomatic patients, and must be differentiated from
other causes of vortex keratopathy (e.g. drug deposition in the cornea). This clouding does not affect vision.
Other ocular findings can include conjunctival and retinal vascular abnormalities, and anterior/posterior spoke-like
cataract. Visual reduction from these manifestastions are uncommon_
Other manifestations
Fatigue, neuropathy (in particular, burning extremity pain), cerebrovascular effects leading to an increased risk of
stroke, tinnitus (ringing in the ears), vertigo, nausea, inability to gain weight, chemical imbalances, and diarrhea
are other common symptoms.
DERMATOLOGICAL FINDINGS
Fabry disease
Corneal verticillata, commonly seen in
patients with Fabry disease, detectable
by slit lamp examination.
DERMATOLOGICAL FINDINGS
DERMATOLOGICAL FINDINGS
DERMATOLOGICAL FINDINGS
•
•
•
Cutaneous lesions in Fabry's
disease:
(A) palms,
(B) lips,
•
(C) labial mucosa
Fabry disease
N Engl J Med 2003; 349
•
Angiokeratomas that were particularly
prominent in the groin and on the
back .(A) Examination of the optic fundi
showed tortuous retinal arterioles (Panel
B). The diagnosis of Fabry's disease
was confirmed by the detection of
reduced α-galactosidase A activity in
peripheral leukocytes . Renal biopsy
showed an accumulation of toluidine
blue staining in cellular inclusions that
was prominent in glomerular podocytes
and also present in distal tubular
epithelial cells and extraglomerular
vascular cells (Panel C, ×80). Electronmicroscopical examination of the
glomerulus revealed podocytes bearing
electron-dense lamellar bodies that
represented secondary lysosomes
containing glycolipid (Panel D, ×3800)
.The patient is being treated with αgalactosidase A administered by
intravenous infusion every two weeks.
Fabry disease
•Fabry's disease:
DERMATOLOGICAL FINDINGS
•
•
•
•
The most common sign reported by patients was reduction of sudoresis. It is frequently presented
in children or adolescents with dry skin and it is translated into intolerance to heat and exercise,
and sometimes into fever without apparent cause . It is suggested that hypohidrosis or anhidrosis
is secondary to selective damage of autonomic nerves, deposits of GL-3 on sweat glands and/or
ischemia owing to impairment of blood vessels that nourish both autonomic nerves and sweat
glands . There are reports of patients with FD that have hyperhidrosis, especially palmar-plantar,
which seems to be more common than previously believed, and it is more prevalent in women .
Body hair may be affected in FD as diffuse body hypotrichosis, by direct built-up of GL-3 in hair
follicles, and also caused by vascular abnormalities of irrigation .
Other common affection detected in the dermatological physical examination is the presence of
edema of eyelids and extremities . Lymphaedema of the legs is common, and it is produced by
GL-3 deposits in the lymphatic vessel . It is rarely responsible for onset of recurrent stasis ulcers,
which might be initial manifestations of the disease .
Fabry disease is suspected based on the individual's clinical presentation, and can be diagnosed
by an enzyme assay (usually done on leukocytes) to measure the level of alphagalactosidase activity. An enzyme assay is not reliable for the diagnosis of disease in females due
to the random nature of X-inactivation. Molecular genetic analysis of the GLA gene is the most
accurate method of diagnosis in females, particularly if the mutations have already been identified
in male family members. Many disease-causing mutations have been noted. Kidney biopsy may
also be suggestive of Fabry disease if excessive lipid buildup is noted.
NON-DERMATOLOGICAL FINDINGS
NEUROPATHIES
•
Neurological: The key symptom of FD is chronic paresthesia,
which occurs especially in homozygote patients, affects hands
and feet, starts in infant years and persists until adult life. It is
burning pain followed by tingling, which can be intermittent or
continuous, and can radiate to neighboring areas .
Acroparesthesias are the main cause of morbidity of the
disease during the two first decades of your life, and in some
cases, they lead to depression and even to suicide attempts .
The pain can be interrupted by the so-called "Fabry crises",
which are episodes of acute pain that last from minutes to
days, which may be followed by fatigue, low grade fever and
arthralgia . They can be triggered by stress, fatigue, disease,
and external temperature increase and/or physical exercise .
Acroparesthesias and crises tend to disappear with time,
possibly owing to complete destruction of nerves fibers . In
adult age, the occurrence of cerebral vascular accident (CVA)
is on average at 34 years of age in homozygote and 40 in
heterozygote subjects, and it is essentially caused by
occlusion of microvasculature or embolism . Cerebrovascular
diseases indicate poor prognosis and, together with
cardiovascular affections, they represent the main causes of
death of these patients . Other neurological affections include
auditory affections (hearing loss), sensorial and vestibular
affections (vertigo and tinnitus) .
•
NEUROPATHIES: PAINFUL
Fabry disease:
Peripheral nerve
Loss of unmyelinated
axons
Electron microscopy for kidney biopsy in Fabry disease
•
Renal: Presence of polyuria
because of defective concentration
may be an early manifestation of
renal impairment, but it is normally
ignored. Most of the patients with
classical forms develop proteinuria
in late adolescent years, and this is
the moment that renal damage is
actually recognized . It is
progressive, normally leads to
chronic renal failure, which is
manifested between the third to fifth
decades of life and it is treated with
chronic dialysis or with renal
transplantation . After the
transplantation, graft enzyme activity
manages to metabolize GL-3
avoiding impairing the transplanted
kidney. Logically, heterozygote
women are not candidates to
donation . Before that, when dialysis
and renal transplantation were not
performed, chronic renal failure was
the main cause of death in FD and
life expectancy was 41 years . In the
so-called renal variant of FD,
patients develop failure in similar
ages to those that have the classical
phenotype, but in the absence of
other disease manifestations
Electron microscopy for kidney biopsy
•
Lipid deposits are characterized
by electron microscopy and
may be seen in different renal
cell types: podocytes,
endothelial glomerular cells,
and tubular cells, primarily
distal. These deposits may be
cleared with ERT(enzyme
replacement therapy); however,
clinical evolution depends
mainly on the presence of
associated renal lesions, such
as tubulo-interstitial fibrosis
and/or glomerulosclerosis. This
photo shows abundant lipid
deposits in podocytes of a 17year-old female Fabry patient
with proteinuria of 500 mg/24 h
(kindly provided by Roser Torra,
Fundació Puigvert, Barcelona)
NON-DERMATOLOGICAL FINDINGS
Ophthalmological changes in Fabry
disease
•
Ophthalmological: There are conjunctival
abnormalities that consist of vascular dilations
and tortuosities that are particularly evident in
the inferior bulbar conjunctiva . Retinal
abnormalities are similar to the previous ones
and can be exacerbated by the presence of
renal disease and high blood pressure, which
is a very common finding in homozygote .
Lens lesions are less frequent and consist of
built-up of granular material and anterior
subcapsular cataract . The most common
ocular findings of FD is cornea verticilata
(yellowish opacities characterized by one or
more lines radiating from a point close to the
corneal core), present in almost all
homozygote subjects and in about 70% to
90% of heterozygote subjects. This affection
does not impact sight. Given that this ocular
finding is very frequent, slit lamp
ophthalmoscopy is an important tool to
support the diagnosis of women with FD when
there is no access to molecular studies . There
may be also xerophthalmia of variable grade
because of reduction of tear production
Ophthalmological changes in Fabry disease
Cornea verticillata
Retinal vessels: Tortuous
NON-DERMATOLOGICAL FINDINGS
•
.
•
Cardiological: Heart impairment is constant in FD. It is caused by GL-3 deposits in the myocardium, in valves
and in the conduction system, which explains the diversity of symptoms, whose expression depends on gender
and age . The most common manifestations are: left ventricle hypertrophy (LVH), mitral failure, arrhythmia, and
arterial coronary disease . LVH that coexists with valve abnormalities are associated with the most severe form of
the disease and are important prognostic factors of severity in general .
Some subjects with low residual levels of enzyme α-GAL develop a variant of the disease that has only heart
expression, named cardiac variant of FD. It is estimated that 3-6% of men with non-obstructive hypertrophic
cardiomyopathy, considered idiopathic, have this FD variant .
Another less known manifestation of this disease is lipid profile abnormality that consists of slightly increased total
cholesterol, HDL cholesterol increase, and normal levels of LDL cholesterol and triglycerides .
.
Gastrointestinal: Episodes of diarrhea, vomiting, post-prandial pain and malabsorption are the most common
complaints and are especially disabling in children. Other findings are pancreatic failure, jejunal diverticuli and
achalasia. These abnormalities are due to accumulation of GL-3 in intestinal blood vessels and autonomic
ganglia .
•
•
•
•
•
Face: Some patients have facial dysmorphy of different grades. The findings are: prominent ear lobes, thick
eyebrows, depressed forehead, prominent nasal angle, large nose, prominent supraorbital bridge, widened nasal
basis and unclear features .
•
Stomatological: Oral cavity findings, in addition to AK, include: xerostomia (by reduction of salivary secretion),
erythema and edema of fungiform papillae, glossitis, en-block tongue, granulomatous keilitis, and increase in
prevalence of cysts and pseudocysts of maxillary sinuses
.
Psychiatric: Depression, suicidal ideation and dementia.
Others: presence of AK in the respiratory and digestive tract mucosa , greater incidence of subclinical
hypothyroidism, airway obstruction, osteopenia up to osteoporosis, anemia, and others .
•
•
•
•
Fabry disease
HISTOPATHOLOGY
• In biopsy of different tissues of FD patients,
histopathological findings consist of the presence of
cytoplasmatic vacuoli that contain accumulated lipids
(previous preservation of tissues by freezing or fixating it
in calcium formol 1%) under light microscopy . Under
electron microscopy we can see the presence of
lysosome inclusions, with typical concentric lamelar
configuration (alternating clear and dark bands each 4-6
mm), called zebra-similar inclusion bodies . When these
findings are not conclusive, we can resort to
immunoelectron microscopy with anti-GL antibodies .
•
Scrotal angiokeratoma
•
Scrotal angiokeratoma; visible large
dilated blood vessels
and hyperkeratosis.
multiple papules made by
dilatated capillaries
Scrotal angiokeratoma
•
Scrotal angiokeratoma (Fordyce type); dilated cavernous capillaries, acanthosis
Genetics
•
•
•
•
•
•
Fabry disease is caused by mutations in the GLA gene. This gene provides instructions for making an enzyme
called alpha-galactosidase A. This enzyme is active in lysosomes, which are structures that serve as recycling
centers within cells. Alpha-galactosidase A normally breaks down a fatty substance called globotriaosylceramide.
Mutations in the GLA gene alter the structure and function of the enzyme, preventing it from breaking down this
substance effectively. As a result, globotriaosylceramide builds up in cells throughout the body, particularly cells
lining blood vessels in the skin and cells in the kidneys, heart, and nervous system. The progressive accumulation
of this substance damages cells, leading to the varied signs and symptoms of Fabry disease.
GLA gene mutations that result in an absence of alpha-galactosidase A activity lead to the classic, severe form of
Fabry disease. Mutations that decrease but do not eliminate the enzyme's activity usually cause the milder, lateonset forms of Fabry disease that affect only the heart or kidneys.
Read more about the GLA gene.
How do people inherit Fabry disease?
This condition is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that
causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who
have only one X chromosome), one altered copy of the GLA gene in each cell is sufficient to cause the condition.
Because females have two copies of the X chromosome, one altered copy of the gene in each cell usually leads to
less severe symptoms in females than in males, or rarely may cause no symptoms at all.
Enzymatic defect of FD leads to accumulation of uncleaved glycosphingolipid (particularly GL-3) in plasma and in
lysosome of innumerous cell types: endothelial cells, pericytes, smooth cells in blood vessels, cardiac myocytes,
epithelial cells of renal glomeruli and tubules, ganglionary cells of autonomic nervous system, corneal cells,
histiocytes and connective tissue cells . GL-3 deposits in endothelial cells lysosome, pericytes and blood smooth
muscle cells produce protuberances of the vessel lumen that cause narrowing and dilations that progress to
ischemia and infarction , as well as accumulation in other type of cells that lead to increase in cell size with onset
of organo -megaly and visceral dysfunction . This pathophysiological mechanism explains the multisystem nature
of this disease
GENETICS
•
•
•
•
FD heritage is X-linked. The affected gene is named GALA
and is found in the region Xq22.1 of chromosome X. GALA is
about 12 kb long and contains about 7 exons that range from
92 to 291 base pairs . The genetic defect that produces the
disease is extremely heterogeneous and current more than
300 mutations have been identified . Most families have
"private mutations", that is, specific mutations to a specific
family .
In male homozygote patients, Fabry gene has high
penetration and most have the classical phenotype of the
disease. In female heterozygous patients, the disease has
variable expressiveness because of the random inactivation of
one X chromosome (Lyon hypothesis). Currently,
heterozygous are not considered simple carriers, given that
most have systemic abnormalities related to the disease and
increase of morbidity and mortality. Therefore, FD
transmission of X-linked recessive form should be
disregarded .
The correlation grade between genotype and phenotype of the
disease is reason for controversy. For example, different
mutations are associated with similar phenotypes, whereas
family members that shared the same mutation have different
phenotypes .
If we suspect of the diagnosis of FD, the family history
investigation is highly important, given that most cases occur
as hereditary. However, absence of positive family data does
not invalidate the diagnosis, given that de novo mutations
have been documented
Investigations
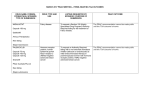
• Investigations
• Confirmation of diagnosis is by demonstration of absent
or deficient levels of alpha-galactosidase A in leukocytes,
plasma or cultured fibroblasts.
• Glycosphingolipid is deposited in urine in complexes.
Urine microscopy with polarised light may show a
'Maltese cross' appearance (birefringent lipid molecules).
• Prenatal diagnosis by enzyme activity or DNA testing in
chorionic villi or cultured amniotic cells is considered in
male fetuses. Pre-implantation diagnosis is possible.
• Other helpful investigations include ECG, MRI,
echocardiography, etc. Eye examination may show
diagnostic corneal or lenticular deposits.
•Pathological finding of Fabry disease
DERMATOLOGICAL DIFFERENTIAL DIAGNOSIS
• Differential diagnosis of isolated AK is made with:
vulgaris wart, senile or rubi angioma, pyogenic
granuloma,Spitz nevus, pigmented seborrheic keratosis,
and melanoma . They should also be differentiated from
other types of AK .
• Disseminated AK is strongly suggestive of FD, but they
are not pathognomonic. For this reason, we should
exclude a broad range of lysosome storage diseases, in
addition to a form of AK corporis diffusum not associated
with known enzymatic abnormalities, called idiopathic or
cutaneous variant of AK corporis diffusum.
DERMATOLOGICAL DIFFERENTIAL DIAGNOSIS
Diagnostic considerations of Fabry disease
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Patients with Fabry disease seek care from a variety of specialists, usually because of the involvement of a
number of organ systems.
The diagnosis and treatment of Fabry disease can be challenging. The signs and symptoms of Fabry disease may
be nonspecific, and if manifestations in different organs are considered in isolation, the unifying diagnosis may be
missed.
The National Society of Genetic Counselors recommends testing for any patient with a family history of Fabry
disease or corneal verticillata ("whorls") on slit lamp exam. In the absence of these factors, it is recommended to
test patients who have any of the following two features:
@Decreased sweating (anhidrosis or hypohidrosis)
@Reddish-purple skin rash in the bathing trunk area (angiokeratomas)
@Personal and/or family history of kidney failure
@Personal or family history of "burning" or "hot" pain in the hands and feet, particularly during fevers
(acroparesthesias)
@Personal or family history of exercise, heat, or cold intolerance
@Patients with sporadic or non-autosomal dominant (no male-to-male) transmission of unexplained cardiac
hypertrophy
If the family history suggests a diagnosis of Fabry disease, genetic testing and counseling should be offered to all
family members, regardless of their sex.
The presence of Fabry symptoms in boys and girls of any age is a strong indication for treatment initiation.
Recommendations from the Fabry Pediatric Expert Panel include the following:
-Treatment should be initiated before irreversible end-organ damage has occurred.
-Asymptomatic children with Fabry mutations should be followed closely.
-The management of Fabry disease requires a multidisciplinary care approach.
MANAGEMENT
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
The treatment of this complex disease requires a multidisciplinary team formed by clinicians, dermatologists, neurologists, cardiologists, nephrologists, and geneticists, all
experienced in the topic.
a. Non-specific therapy:
It is support care, directed only to controlling the symptoms and signs present and it normally complements specific therapy.
Angiokeratomas: may be destroyed by different methods: electrocoagulation, cryotherapy, surgical exeresis or laser (Neodymium-YAG, pulsed dye, and others). Laser
therapy is the therapy of choice .
Acroparesthesias, Fabry crisis: We ask the patients to identify and avoid predisposing symptoms. Partial improvement of pain is obtained with diphenylhydantoine,
carbamazepine, gabapentine, oxacarbazepine or topiramate
Cerebral and retinal vascular disease: prevention is made with antineoplastic or anti-coagulant agents. Vascular protection may be enhanced with inhibitors of
angiotensin conversion enzyme, statins and folic acid5,.
Renal disease: control of arterial hypertension, dialysis, and even renal transplant . Aggressive treatment is indicated with angiotensin converting enzyme inhibitor or with
blockers of angiotensin receptors to reduce proteinuria
Cardiac disease: control of arrhythmia with anti-arrhythmia drugs, pacemakers (if indicated), up to heart transplantation. The patients with coronary disease can be
candidates to coronary revascularization .
b. Specific Therapy:
Enzymatic replacement therapy (ERT)
This strategy of treatment is based on the discovery that cells can incorporate an enzyme into the extracellular medium and use it for its normal metabolism. ERT for FD
was approved in Europe in 2001 and in the United States in 2003. Currently, there are two human α-GAL available in the market: alpha algasidase (Replagal®,
Transkaryotic Therapies Inc., Cambridge, Massachusetts), produced by human fibroblast cultures added by active promoters for gene transcription of α-GAL, approved in
Europe, and beta algasidase (Fabrazyme®, Genzyme Corp., Cambridge, Massachusetts), obtained from recombinant therapy of hamsters' ovaries, approved in Europe
and USA 5,68. Both proteins are structurally and functionally similar, without specific comparable activity and are administered by intravenous via every 15 days. Doses
are variable depending on the preparation: 0.2 mg/kg/dose of alpha algasidase and 1 mg/kg/dose of beta algasidase.
ERT is a treatment to be used for their entire life, given that the amount of plasmatic enzyme is rapidly depleted, requiring constant infusions 68. Tolerance to ERT is
normally good, except for mild or moderate reactions associated with infusion, product of formation of non-neutralizing IgG antibodies.
It is clear that ERT reverses metabolic abnormalities and many pathological affections of FD. The purpose of implementation is to prevent the development of the disease
in young patients and to avoid or reverse the progression of organic dysfunction in older patients .
Homozygote:
- below 16 years: as soon as there are signs and symptoms
- over 16 years: upon diagnosis
Heterozygote: when there are significant symptoms or key organ impairment.
Clinical trials with both enzymatic preparations show reduction in frequency of pain episodes, heart masses, and GL-3 deposits in kidneys (with stabilization or
improvement of renal function in cases of mild impairment) and on the skin. There is evidence that ERT improves sudoresis, gastrointestinal symptoms, pulmonary and
auditory disorders . However, there is no information about the long-term impact of ERT in FD mortality.
Gene therapy: This technique aims at adding normal gene of α-GAL to normal gene of the patient's DNA, which starts to producing normal enzyme. The methods used to
introduce foreign genes in cells are basically classified as viral systems (they use viral vectors such as oncoretroviruses, lentiviruses or adenoviruses) and non-viral
systems (liposome containing DNA). Both delivery systems have been tested in FD. Gene therapy proposes definite cure of the disease, but it is still at experimental
stage .
FD is a severe multisystemic disease. Current availability of specific treatment forces us to make the diagnosis as early as possible. Dermatologically speaking, we should
recognize that AK and hypohidrosis are the two signs that support the diagnosis
Enzyme replacement therapy (ERT)
•
Current treatment for Fabry disease
involves enzyme replacement therapy
(ERT) with intravenous infusions of
recombinant human α-galactosidase
A.
•
•
This therapeutic regimen consistently
decreases Gb3 levels in plasma and
clears lysosomal inclusions from
vascular endothelial cells.
•
•
The effects of ERT on other tissues
are not as obvious, suggesting that
treatment must be initiated early in the
course of the disease to be optimally
effective or that some complications of
the disease are not responsive to
enzymes delivered intravenously
Related disorders to Fabry disease
•
•
•
•
•
•
Gaucher disease is one of the most common of the lipid storage diseases and is characterized by the abnormal accumulation of
certain fatty substances in various organs of the body. Symptoms develop due to a deficiency in the enzyme glucocerebrosidase and may
include enlargement of the liver (hepatomegaly) and spleen (splenomegaly), a general feeling of ill health (malaise), visual difficulties,
abdominal swelling, severe bone pain and bone disease. Gaucher disease is inherited as an autosomal recessive trait.
Fucosidosis is an extremely rare inherited lysosomal storage disease characterized by a deficiency of the enzyme alpha-L-fucosidase.
There are at least two types of fucosidosis (i.e., type 1 and type 2), determined mainly by the severity of the enzyme deficiency and
resulting symptoms. The symptoms of fucosidosis type 1, the most severe form of the disease, may become apparent as early as six
months of age. Symptoms may include a skin lesion similar to Fabry disease (angiokeratoma), progressive deterioration of the brain and
spinal cord , intellectual disability, loss of previously acquired intellectual skills, and growth retardation leading to short stature. Other
physical findings and features become apparent over time, including multiple deformities of the bones (dysostosis multiplex), coarse facial
features, enlargement of the heart (cardiomegaly), enlargement of the liver and spleen (hepatosplenomegaly), and/or episodes of
uncontrolled electrical activity in the brain (seizures). Additional symptoms may include increased or decreased perspiration and/or
malfunction of the gallbladder and/or salivary glands. Fucosidosis is inherited as an autosomal recessive trait.
Schindler disease is a rare inherited metabolic disorder characterized by a deficiency of the lysosomal enzyme alpha-Nacetylgalactosaminidase (alpha-NAGA), which leads to an abnormal accumulation of certain complex compounds (glycosphingolipids and
oligosaccharides) in many tissues of the body. Schindler disease is inherited as an autosomal recessive disorder. There are three types of
Schindler disease.
The classical form of the disorder, known as Schindler disease, type I, has an infantile onset. Affected individuals appear to develop
normally until approximately one year of age, when they begin to lose previously acquired skills that require the coordination of physical
and mental activities (developmental regression). Additional neurological and neuromuscular symptoms may become apparent, including
diminished muscle tone (hypotonia) and weakness; involuntary, rapid eye movements (nystagmus); visual impairment; and episodes of
uncontrolled electrical activity in the brain (seizures). With continuing disease progression, affected children typically develop restricted
movements of certain muscles due to progressively increased muscle rigidity, severe intellectual disability, hearing and visual impairment,
and a lack of response to stimuli in the environment.
Type 2 Schindler disease also known as Kanzaki disease, is the adult-onset form with symptoms presenting in the second or third decade
of life. The disorder is characterized by angiokeratoma, a skin lesion and distribution similar to that seen in type 1 classic Fabry disease.
Presentation may also include lymphedema, intellectual impairment, and distinct facial features including mildly coarse features, thick lips,
a depressed nasal bridge and an enlarged tip of the nose.
Type III Schindler disease is an intermediate form the disorder. Symptoms can range from more serious intellectual impairment,
neurological dysfunction and seizures to milder neurological and psychiatric issues such as speech and language delays and mild autismlike symptoms.
Types of angiokeratoma
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Symptoms of Angiokeratoma
Depending on the type of angiokeratoma, the patient can present any of the following symptoms:
Angiokeratoma of Mibelli (telangiectatic warts):
Red papules on the skin
Size of 1-5 mm
Skin becomes thicker in time.
Angiokeratoma of Fordyce (angiokeratoma of the scrotum and vulva):
Red or blue papules on the scrotum or vulva (labia majora)
Can also affect the shaft of the penis, the inner thigh and the lower abdomen
Single or multiple lesions (over 100)
In young people, lesions are small in size, of red color and with scales
In older people, lesions are large in size, of blue or black color and the scale tend to overlap.
Many do not present any symptoms but they are noticed after the patient scratches the skin or has intercourse (bleed).
Solitary angiokeratoma:
Wart-like papules on the skin
Small size
Color blue or black
Present on lower extremities.
Angiokeratoma circumscriptum naeviforme (verrucous vascular malformation):
The subcutaneous capillaries and veins present a malformation
Verrucous component
Telangiectatic lesions, the appear clustered in a small area, most often on the leg or trunk
Their color becomes darker with time and they can also change their size and shape
Acanthosis and hyperkeratosis
Solitary lesion in rare cases.
Angiokeratoma corporis diffusum (Fabry syndrome):
Lesions are widespread on the lower trunk and groin area
Fever
Painful hands and feet
Requires emergency medical attention and treatment.
Angiokeratomas are small dark red to purple raised spots. They may also have a rough scaly
surface. They are composed of surface blood vessels (dilated capillaries). Often unnoticed, they may
become crusty and bleed if accidentally scratched or damaged, or a harmless clot may form in the
lesion (thrombosis), changing the color to dark purple or black overnight
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
.
Type of angiokeratomaDescription
Sporadic angiokeratoma
Solitary lesions
Common in those over 40 years
Angiokeratoma of Fordyce
Most commonly found on the scrotum. Also found on the shaft of the penis, labia majora of the vulva, inner thigh and lower abdomen
Most prevalent in those over 40 years
Men are more often affected than women
May be a single lesion or multiple lesions (>100)
Lesions are small, red and less scaly in younger patients whilst in older patients they tend to be larger, blue/black and with overlying
scales
Usually symptomless and may only be noticed when they bleed after scratching or intercourse
Angiokeratoma circumscriptum
Rare birthmark (vascular malformation). May be present at birth but can occur later in childhood or adulthood
Females are more affected than men 3:1
Cluster of lesions on a small area of the leg or trunk
Over time lesions may darken in colour and change shape and size
Fabry syndrome (angiokeratoma corporis diffusum)
Rare serious inherited disorder caused by a deficiency of an alpha-galactosidase enzyme, ceramide trihexosidase
Excessive quantities of glycosphingolipids are deposited in blood vessels and internal organs
More severe in males than females
Angiokeratomas are widespread, most numerous on lower trunk and groin area
May present with fever and painful hands & feet
May result in corneal opacities, kidney failure, heart failure, strokes, arthritis, colitis & many other problems.
Angiokeratoma
Pathology outline, coms 2O12
•
Vascular lesion characterized by
superficial vascular ectasia and
overlying acanthosis or
hyperkeratosis
● Solitary or diffuse
Five types with similar histology:
(a) angiokeratoma of Mibelli – acral
skin
(b) angiokeratoma of Fordyce –
scrotal skin
(c) angiokeratoma corporis
diffusum – seen in Fabry disease
(d) angiokeratoma circumscriptum
– usually congenital, associated
with nevus flammeus, cavernous
hemangioma
(e) solitary or multiple
angiokeratomas
Angiokeratoma
corporis naeviform
•
Angiokeratomas are a group of vascular
ectasias that involve the papillary dermis
and may produce papillomatosis,
acanthosis, and hyperkeratosis of the
epidermis. Several clinical variants of
angiokeratomas exist; angiokeratoma
circumscriptum is one type, and the least
frequent of the other types of
angiokeratomas. Overall, 8 types of
angiokeratomas have been described in
the literature. The first reported case
dates as far back as 1889 when Mibelli
described what is now known as
angiokeratoma Mibelli-type on the
fingers and the toes. Fabry first
described angiokeratoma
circumscriptum in 1915 as a localized
lesion on a lower extremity or the trunk.
In addition, a rare manifestation of
angiokeratoma circumscriptum
naeviforme, with appearance on the
neck, has been documented. These
lesions are of clinical importance
because, often being dark purple or
black in nature, they may clinically mimic
a malignant melanoma Angiokeratoma
circumscriptum has also been called
angiokeratoma corporis naeviform and
may be best classified as a type of
capillary malformation.
Angiokeratoma circumscriptum naeviforme
Dermatology Online Journal 17 (9): 11
Angiokeratoma circumscriptum
neviforme: Year :Ind. Derm.
Online J. 2014 : 5 : 4 : 472
Angiokeratoma Solitary angiokeratoma
Angiokeratoma circumscriptum of the tongue
J. Cut. & Aesthetic Surg.: 2011 : 4 : 3 : 205
•
•
•
•
•
•
•
•
Angiokeratomas can be divided into localized
and systemic forms.
The localized forms include the following:
(i) solitary papular angiokeratoma that typically
occurs on the lower extremities,
(ii) localized angiokeratoma of the scrotum and
vulva (Fordyce type),
(iii) congenital form, AC naeviforme that
presents as multiple hyperkeratotic papular and
plaque-like lesions, usually unilaterally on the
lower leg, foot, thigh, buttock or occasionally
elsewhere and
(iv) bilateral angiokeratomas that occur on the
dorsa of the fingers and toes (Mibelli type).
Generalized systemic form, angiokeratoma
corporis diffusum, is usually associated with a
metabolic disorder, the most common are
Fabry's disease and fucosidosis.
Mucosal involvement, including the oral cavity,
has been described both in systemic forms and
as a component of localized ones. However,
solitary angiokeratomas of the oral mucosa, not
associated with lesions elsewhere, are rare and
only few cases of isolated mucosal
angiokeratomas have been reported.
Angiokeratoma
Eyelid angiokeratoma
MEAJO: 2014 : 21 : 3 : 287
Angiokeratoma
Angiokeratoma circumscripta
Angiokeratoma
circumscriptum
Ped. Derm.2015 : 16 : 3 : 165
Angiokeratoma circumscriptum
2O14: 57 : 2 : 350
Angiokeratoma (Mibelli type)
•
•
•
•
Angiokeratomas are vascular
malformations, which are clinically
characterized by solitary or multiple
papules or plaques. The lesion
histologically defined as dilated blood
vessels with sub epidermal position
and it has epidermal proliferative
reaction. In all form of angiokeratoma,
the epidermal changes occur in the
secondary stage.
There are several clinical types of
angiokeratoma depending on the
multiplicity and location of the
lesions. Angiokeratoma corporis
diffusum is the systemic form, which
frequently link to some inborn error of
metabolism, primarily Fabry's disease
and fucosidosis.
The localized forms are;
(1) solitary papular angiokeratomas;
(2) scrotal or vulval angiokeratomas;
(3) multiple congenital
angiokeratomas; and
(4) bilateral angiokeratomas in the
dorsum of hands and feet (Mibelli
type).
Scrotal angiokeratoma (Mibelli type); blood vessels close to the epidermis
Angiokeratoma of the scrotum
•
Angiokeratoma
scrotale (Fordyce)
Vulvar angiokeratomas
(Fordyce)
•
Verrucus hemangioma
•
来源 生物谷
•
Boonnews 2007, 2:41:23