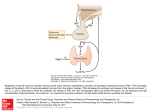
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
University of Iowa Iowa Research Online Theses and Dissertations Spring 2013 Investigating the genetics Of thyroid stimulating hormone in newborns Farah Yacoub Alul University of Iowa Copyright 2013 Farah Yacoub Alul This dissertation is available at Iowa Research Online: http://ir.uiowa.edu/etd/2434 Recommended Citation Alul, Farah Yacoub. "Investigating the genetics Of thyroid stimulating hormone in newborns." PhD (Doctor of Philosophy) thesis, University of Iowa, 2013. http://ir.uiowa.edu/etd/2434. Follow this and additional works at: http://ir.uiowa.edu/etd Part of the Genetics Commons INVESTIGATING THE GENETICS OF THYROID STIMULATING HORMONE IN NEWBORNS by Farah Yacoub Alul An Abstract Of a thesis submitted in partial fulfillment of the requirements for the Doctor of Philosophy degree in Genetics in the Graduate College of The University of Iowa May 2013 Thesis Supervisor: Professor Jeffrey C. Murray 1 ABSTRACT Endocrine disorders are substantial contributors to neonatal morbidity and mortality and, of these, congenital hypothyroidism (CH) is the most common (Kumar et al. 2009). CH is a common and preventable cause of mental retardation with an incidence of approximately 1 in 2,350 live births (Hinton et al. 2010). In Iowa, the Iowa Neonatal Metabolic Screening program (INMSP) uses thyroid stimulating hormone (TSH) to screen for CH at birth. However, TSH is highly variable among healthy newborns as well as adults, leading to false positive results in some cases. Previous studies have observed that adult TSH variability is under strong genetic regulation with estimated heritability of up to 65% (Panicker et al. 2008). Additionally, there have been multiple studies examining genetic factors associated with adult TSH levels. TSH heritability has never been estimated in the neonatal period and we aimed to determine the heritability in neonates and compare it to heritability estimates in adults. We examined 381 twin pairs obtained from the INMSP. Heritability was estimated using multilevel mixed-effects linear regression adjusting for factors affecting TSH levels; gestational age, gender, weight and age at time of sample collection. We estimated neonatal TSH heritability to be 58% with a P-value of 2x10-5, which mirrors adult heritability estimates, and provides direct evidence for a strong genetic contribution to TSH variability at birth. We next examined genetic factors that may contribute to the observed heritability. Genetic contribution to TSH variation has been studied extensively in adults, but not in neonates. We genotyped a population of Iowa neonates; term (n=827) and preterm (n=815), for 45 single nucleotide polymorphisms (SNPs) that we selected based on reported genetic associations with adult TSH levels from the literature, as well as other candidate genes. TSH values were obtained from the INMSP. Analysis of variance was performed to identify genetic associations with TSH concentrations. The strongest 2 association identified was rs4704397 in the PDE8B gene (p=1.3x10-4), followed by rs965513 (p=6.4x10-4) on chromosome 9 upstream of the FOXE1 gene. Both of these SNPs met statistical significance after correction for multiple testing. Our results demonstrated for the first time two genetic associations with neonatal TSH levels that replicate findings with adult TSH levels, and these findings could have clinical implications for the early prediction of risk for adult diseases and conditions associated with thyroid hormone levels. Finally, we aimed to identify the etiologic variants that may be responsible for the observed associations by fine mapping adjacent enhancer elements. This was done in two stages; first we sequenced 58 term neonates with TSH levels at both ends of the normal distribution to identify associated variants, then we replicated the findings in an additional population of 306 term neonates. None of the observed variants were statistically significant for an association with TSH levels, however, one of the rare variants (rs112053411) identified was detected in three neonates, all in the upper distribution of TSH levels including one infant that was later diagnosed with CH. This rare variant is worth pursuing in a larger study population and in functional studies. Together this work has advanced our knowledge of the genetic basis of TSH variation in the neonatal period, as well as provided insight into the early prediction of risk for adulthood thyroid related diseases through shared genetic associations between adults and neonates. Abstract Approved: ____________________________________ Thesis Supervisor ____________________________________ Title and Department ____________________________________ Date INVESTIGATING THE GENETICS OF THYROID STIMULATING HORMONE IN NEWBORNS by Farah Yacoub Alul A thesis submitted in partial fulfillment of the requirements for the Doctor of Philosophy degree in Genetics in the Graduate College of The University of Iowa May 2013 Thesis Supervisor: Professor Jeffrey C. Murray Copyright by FARAH YACOUB ALUL 2013 All Rights Reserved Graduate College The University of Iowa Iowa City, Iowa CERTIFICATE OF APPROVAL _______________________ PH.D. THESIS _______________ This is to certify that the Ph.D. thesis of Farah Yacoub Alul has been approved by the Examining Committee for the thesis requirement for the Doctor of Philosophy degree in Genetics at the May 2013 graduation. Thesis Committee: ___________________________________ Jeffrey C. Murray, Thesis Supervisor ___________________________________ Alexander G. Bassuk ___________________________________ Terry A. Braun ___________________________________ Josep M. Comeron ___________________________________ John M. Dagle To my beloved family. ii ACKNOWLEDGMENTS There are so many people that have helped and supported me and without whom this work would not have been possible. First I would like to thank Dr. Jeff Murray for giving me this amazing opportunity. When I first came to Iowa I was very fortunate to work as a research assistant in Dr. Murray’s lab and that paved the way for me to enter the Genetics Program. Dr. Murray goes above and beyond for his students and no one can wish for a better mentor. I would also like to thank Dr. Kelli Ryckman. Kelli was like a second mentor to me and I learned so much from her, as well as a great friend. I am sure I would not be where I am today if it weren’t for Kelli’s support. I would like to thank the members of my thesis committee: Dr. John Dagle, Dr. Alex Bassuk, Dr. Terry Braun, and Dr. Josep Comeron for valuable insight and advice in every meeting, and Dr. Anne Kwitek for her role in my comprehensive exam. I am also thankful for the support of Dr. Dan Eberl, Anita Kafer, Isabelle Hardy, and Linda Hurst from the Genetics Program. I was very fortunate to work in one of the biggest labs on campus, so there are so many people to thank. I would like to start with Tamara Busch for answering my never ending questions, Elizabeth Leslie and Leah Biggs for all your help with comps. Thanks to Allison Momany and Lauren Fleener for being great friends and making me laugh every day, you two are the best bench neighbors ever. I am also very grateful to Lauren Fleener, Osayame Ekhaguere and Elwood Cook; without your help I would still be processing blood spot cards today. I am very thankful to all past and present Murray lab members, you all make coming to work every day something to look forward to: Nancy Weathers, Adela Mansilla, Aline and Raul Petrin, Jennifer Standley, Brandon Alleman, Bruce Bedell, Lindsey and Dana Rhea, Azeez Butali, Zhonglin Jia, Hodad Naderi, Jinsil Kim, Aimee Buhr, Satoshi Suzuki, Dee Even, Nancy Davin, Erin Brothers-Smith, Sarah Barten, and Susie McConnell. I would also like to thank my grad school colleagues and iii great friends, Lily Paemka and Dina Ahram, I was fortunate to go through this journey with you. I would like to thank all of the families that participated in our research, without them none of this would be possible. I am also very grateful to people at the State Hygienic Lab especially Dr. Stanton Berberich for providing samples, and the University of Iowa DNA Facility especially Dr. Kevin Knudtson, Mary Boes and Garry Hauser, for providing me with work space, machines and support when results didn’t turn out as expected. I would like to thank Drs. Zuhair Amr and Abdelkader Battah from Jordan, without their guidance I would not have made it to grad school to begin with. And I would especially like to thank Dr. Adel Afifi for introducing me to Dr. Murray when I first came to Iowa, and for being an exceptional supporter of new graduates coming to the United States from abroad. Finally, I am very grateful for my family. I owe everything good that happened to me to my parents Yacoub and Eman Alul, they have been exceptionally supportive and loving my whole life and I wish one day I can be to my children half the parent they were to me. I also want to express my gratitude to my sisters Lubna and Haya, my grandmothers Amal and Nahida, and my aunts May and Azza for the endless support and love. Last but not least, I would like to express my honest gratitude to my husband Taher and my son Bashir. They were with me every step of the way, and witnessed this whole experience with me first hand. Taher’s unconditional love and support was what got me through the ups and downs throughout this journey. He always pushes me for more and expects me to excel. And Bashir always helps me forget any troubled experiment the minute I pick him up from daycare. I owe all of this and much more to you all. iv ABSTRACT Endocrine disorders are substantial contributors to neonatal morbidity and mortality and, of these, congenital hypothyroidism (CH) is the most common (Kumar et al. 2009). CH is a common and preventable cause of mental retardation with an incidence of approximately 1 in 2,350 live births (Hinton et al. 2010). In Iowa, the Iowa Neonatal Metabolic Screening program (INMSP) uses thyroid stimulating hormone (TSH) to screen for CH at birth. However, TSH is highly variable among healthy newborns as well as adults, leading to false positive results in some cases. Previous studies have observed that adult TSH variability is under strong genetic regulation with estimated heritability of up to 65% (Panicker et al. 2008). Additionally, there have been multiple studies examining genetic factors associated with adult TSH levels. TSH heritability has never been estimated in the neonatal period and we aimed to determine the heritability in neonates and compare it to heritability estimates in adults. We examined 381 twin pairs obtained from the INMSP. Heritability was estimated using multilevel mixed-effects linear regression adjusting for factors affecting TSH levels; gestational age, gender, weight and age at time of sample collection. We estimated neonatal TSH heritability to be 58% with a P-value of 2x10-5, which mirrors adult heritability estimates, and provides direct evidence for a strong genetic contribution to TSH variability at birth. We next examined genetic factors that may contribute to the observed heritability. Genetic contribution to TSH variation has been studied extensively in adults, but not in neonates. We genotyped a population of Iowa neonates; term (n=827) and preterm (n=815), for 45 single nucleotide polymorphisms (SNPs) that we selected based on reported genetic associations with adult TSH levels from the literature, as well as other candidate genes. TSH values were obtained from the INMSP. Analysis of variance was performed to identify genetic associations with TSH concentrations. The strongest v association identified was rs4704397 in the PDE8B gene (p=1.3x10-4), followed by rs965513 (p=6.4x10-4) on chromosome 9 upstream of the FOXE1 gene. Both of these SNPs met statistical significance after correction for multiple testing. Our results demonstrated for the first time two genetic associations with neonatal TSH levels that replicate findings with adult TSH levels, and these findings could have clinical implications for the early prediction of risk for adult diseases and conditions associated with thyroid hormone levels. Finally, we aimed to identify the etiologic variants that may be responsible for the observed associations by fine mapping adjacent enhancer elements. This was done in two stages; first we sequenced 58 term neonates with TSH levels at both ends of the normal distribution to identify associated variants, then we replicated the findings in an additional population of 306 term neonates. None of the observed variants were statistically significant for an association with TSH levels, however, one of the rare variants (rs112053411) identified was detected in three neonates, all in the upper distribution of TSH levels including one infant that was later diagnosed with CH. This rare variant is worth pursuing in a larger study population and in functional studies. Together this work has advanced our knowledge of the genetic basis of TSH variation in the neonatal period, as well as provided insight into the early prediction of risk for adulthood thyroid related diseases through shared genetic associations between adults and neonates. vi TABLE OF CONTENTS LIST OF TABLES ............................................................................................................. ix LIST OF FIGURES ........................................................................................................... xi CHAPTER1 INTRODUCTION ..........................................................................................1 Significance ......................................................................................................1 The Thyroid Gland ...........................................................................................2 Thyroid Hormones ............................................................................................3 Thyroid Follicles ...............................................................................................4 Hypothalamic-Pituitary-Thyroid Axis ..............................................................4 Thyroid Stimulating Hormone ..........................................................................5 Embryological Development of the Thyroid Gland .........................................5 Molecular Aspects of Thyroid Morphogenesis ................................................6 Thyroid Function in the Fetus...........................................................................7 Thyroid Function in the Neonate ......................................................................7 Full Term Newborns..................................................................................7 Preterm Newborns .....................................................................................8 Disorders of Thyroid Function .........................................................................8 Transient Hypothyroxinemia of Prematurity.............................................8 Congenital Hypothyroidism ......................................................................9 Newborn Screening ........................................................................................10 Etiology of Congenital Hypothyroidism ........................................................12 Transient Congenital Hypothyroidism ....................................................12 Permanent Congenital Hypothyroidism ..................................................13 Adult Onset Hypothyroidism ..........................................................................14 Genetics of Thyroid Stimulating Hormone ....................................................15 Genome Wide Association Studies .........................................................16 Candidate Gene Studies ...........................................................................19 Main Thyroid Related Genes...................................................................20 Purpose of Study .............................................................................................22 Hypothesis ...............................................................................................23 Specific Aims ..........................................................................................23 CHAPTER 2 THE HERITABILITY OF METABOLIC PROFILES IN NEWBORN TWINS ......................................................................................33 Abstract ...........................................................................................................34 Introduction.....................................................................................................34 Methods ..........................................................................................................37 Study Population .....................................................................................37 Enzymatic Assays ....................................................................................37 Tandem Mass Spectrometry ....................................................................38 Determination of Zygosity ......................................................................38 Statistical Analysis ..................................................................................39 Analysis of Ratios....................................................................................41 Results.............................................................................................................42 Discussion .......................................................................................................43 vii CHAPTER 3 GENETIC ASSOCIATIONS WITH NEONATAL THYROID STIMULATING HORMONE LEVELS ........................................................57 Abstract ...........................................................................................................58 Introduction.....................................................................................................58 Methods ..........................................................................................................60 Study Population .....................................................................................60 Marker Selection and Genotyping ...........................................................61 Statistical Analysis ..................................................................................62 Haplotype Analysis .................................................................................63 Results.............................................................................................................63 Discussion .......................................................................................................65 CHAPTER 4 CHARACTERIZATION OF FOXE1 AND PDE8B ENHANCER REGIONS FOR A ROLE IN NORMAL VARIATION IN NEONATAL TSH LEVELS ..........................................................................83 Abstract ...........................................................................................................83 Introduction.....................................................................................................84 Materials and Methods ...................................................................................86 Study Population .....................................................................................86 Selection of Studied Regions...................................................................88 Genotyping ..............................................................................................88 Statistics ...................................................................................................89 Results.............................................................................................................90 Sequencing ..............................................................................................90 TaqMan....................................................................................................91 Discussion .......................................................................................................91 CHAPTER 5 CONCLUSIONS AND FUTURE DIRCTIONS .......................................101 Conclusions...................................................................................................101 Future Directions ..........................................................................................107 REFERENCES ................................................................................................................110 viii LIST OF TABLES Table 1-1 Summary of GWAS data for TSH.....................................................................24 Table 2-1 List of analytes examined. .................................................................................47 Table 2-2 List of markers used for zygosity testing and population allele frequencies.........................................................................................................48 Table 2-3 Heritability estimates for all twin pairs. ............................................................49 Table 2-4 Heritability estimates for twin pairs excluding TPN, abnormal screens, gestational age <34 weeks, and MZ twin pairs that are >20% discrepant in weight. ...........................................................................................................51 Table 2-5 Demographic characteristics of twin pairs. .......................................................53 Table 2-6 Heritability estimates for analyte ratios. ............................................................54 Table 3-1 List of genotyped markers. ................................................................................70 Table 3-2 Demographic Characteristics of Cohorts...........................................................72 Table 3-3 Association statistics for all genotyped SNPs with neonatal TSH level for all combined populations. P-values reported in this table are from ANOVA and non-parametric analysis. .............................................................73 Table 3-4 Association statistics for all genotyped SNPs with neonatal TSH level for individual population cohorts. P-values reported in this table are from ANOVA and non-parametric analysis. .............................................................75 Table 3-5 TSH means and standard deviations for PDE8B and FOXE1 significant SNPs. .................................................................................................................77 Table 3-6 TSH means and standard deviations for marginally significant SNPs. .............78 Table 3-7 SNP haplotypes in the FOXE1 gene that are significantly associated (p<0.001) with natural log transformed TSH levels..........................................79 Table 3-8 SNP haplotypes in the TSHR gene that are significantly associated (p<0.001) with natural log transformed TSH levels..........................................80 Table 4-1 Potential FOXE1 and PDE8B enhancer regions studied. ..................................95 Table 4-2 Primer sequences and PCR conditions for sequencing FOXE1 and PDE8B enhancer regions...................................................................................96 ix Table 4-3 List of SNPs selected for further investigation after sequencing. .....................97 Table 4-4 List of all sequence variants detected in the sequencing of 58 newborns. ........98 x LIST OF FIGURES Figure 1-1 Thyroid gland. . ................................................................................................25 Figure 1-2 Iodine organification and thyroid hormone synthesis. .....................................26 Figure 1-3 Hypothalamic-pituitary-thyroid axis regulation. ..............................................27 Figure 1-4 Three month old infant with untreated congenital hypothyroidism. . ..............28 Figure 1-5 A Manhattan plot of the TSH level GWAS done in 2008 by ArnaudLopez et al. ......................................................................................................29 Figure 1-6 A Manhattan plot of the TSH level GWAS done in 2010 by Panicker et al. . ...................................................................................................................30 Figure 1-7 A Manhattan plot of the TSH level GWAS meta-analysis done in 2012 by Rawal et al. . ...............................................................................................31 Figure 1-8 A Manhattan plot of the TSH level GWAS meta-analysis done in 2013 by Porcu et al. . ................................................................................................32 Figure 2-1 Heritability of analyzed metabolites and analyte ratios. . ................................55 Figure 2-2 Environment and genetic contribution to metabolites in the β-oxidation pathway and the pathway for catabolism of branched-chain amino acids.......56 Figure 3-1 Association statistics for all genotyped SNPs with neonatal TSH level for combined populations. ...............................................................................81 Figure 3-2 Association statistics for all genotyped SNPs with neonatal TSH level for individual populations. ...............................................................................82 Figure 4-1 FOXE1 potential enhancer regions. .................................................................99 Figure 4-2 PDE8B potential enhancer regions. ...............................................................100 xi 1 CHAPTER1 INTRODUCTION Significance Endocrine disorders are substantial contributors to neonatal morbidity and mortality and, of these, congenital hypothyroidism (CH) is the most common (Kumar et al. 2009). CH is a common and preventable cause of mental retardation with an incidence of approximately 1 in 2,350 live births (Hinton et al. 2010). Early treatment with thyroxine (T4) with subsequent supplementation for life produces excellent results for both growth and development (Wilcken and Wiley 2008; Hinton et al. 2010). CH is screened for at birth through detection of T4, thyroid stimulating hormone (TSH), or both (Wilcken and Wiley 2008). Unfortunately, newborn screening for CH has the highest number of false positive results with repeated testing costs of more than two million dollars annually (Kwon and Farrell 2000). False positive results can cause significant stress for parents of these infants, and also result in an increased financial burden in follow up testing costs (Kwon and Farrell 2000). The false positive result rate is usually higher in newborn screening programs that use T4 as a primary analyte (0.30%) compared to programs that use TSH (0.05%) (Kaye et al. 2006b). In Iowa, the Iowa Neonatal Metabolic Screening program uses TSH only to screen for CH (Pass and Neto 2009). Furthermore, TSH is extremely sensitive to serum thyroid hormone levels, which makes it the test of choice for clinicians to diagnose most cases of thyroid dysfunction (Stathatos 2012). TSH is highly variable among healthy newborns; this is one of the factors contributing to the false positive rate for this test when used in newborn screening programs. Additionally, TSH varies greatly by gestational age and birth weight and shows lower levels in premature or low birth weight newborns (Kirsten 2000). In healthy adult subjects, TSH has considerable variability between individuals, whereas this 2 variability is much less in the same individual when TSH is measured repeatedly over an extended period of time (Panicker et al. 2010). Previous studies have observed that adult TSH variability is under strong genetic regulation; studies have estimated a heritability of up to 65% (Panicker et al. 2008). However, no studies have examined the heritability of neonatal TSH levels. Furthermore, there have been multiple studies examining genetic factors associated with adult TSH levels but no studies have examined the genetics of neonatal TSH levels. Therefore, a better understanding of the normal variation in TSH may improve the sensitivity of its use in newborn screening and help minimize the rate of false positive results, as well as provide a better understanding of the thyroid profile and disorders in newborns. Understanding variation in TSH levels and the genes responsible may be particularly important in a population at risk for abnormal TSH levels such as preterm infants. Preterm infants manifest even higher TSH variability than term infants owing to the postnatal changes in thyroid function due to the premature interrupted exposure to maternal thyroid hormones as well as an immature hypothalamic-pituitary axis (HPA) (Hinton et al. 2010). Thyroid hormones also play a key role in neonatal as well as adult normal physiology, affecting almost all tissues and maintaining healthy status of all human systems including cognition, cardiovascular function, skeletal health, and balanced energy and metabolic status (Panicker 2011). Understanding the shared genetic associations with TSH levels in both the neonatal period as well as through adulthood will be useful for earlier prediction of risk to adult diseases that are affected by TSH levels. The Thyroid Gland The thyroid is a butterfly-shaped endocrine gland located in the neck immediately below the larynx and anterior to the trachea (Figure 1-1) (Kirsten 2000; Stathatos 2012). The thyroid gland consists of two lateral lobes connected by narrow thyroid tissue called 3 the isthmus (Figure 1-1). The two thyroid lobes contain spherical structures called follicles (Kirsten 2000). The thyroid follicle is the functional part of the thyroid gland consisting of the thyroid hormone synthesizing follicular cells (De Felice and Di Lauro 2011; Stathatos 2012). During human embryological development, the thyroid is the first endocrine structure to develop (Kratzsch and Pulzer 2008). It is also one of the largest endocrine glands, weighing about 15 to 20 grams in human adults (Kirsten 2000). The thyroid gland secretes thyroid hormones that are vital for the normal physiology of virtually every organ system in the human body (Kirsten 2000; Panicker 2011). Thyroid Hormones The thyroid gland is necessary for normal human physiology, plays a vital role in almost all tissues, and influences most bodily functions through the secretion of its two major thyroid hormones; thyroxine (T4) and triiodothyronine (T3) (Kirsten 2000; Panicker 2011). Of these two thyroid hormones, T3 is considered to be the primary thyroid hormone since it is more potent, but T4 is more abundant (Kirsten 2000). Under normal conditions, thyroid gland output is about 90% T4 and 10% T3 (Stathatos 2012). Thyroid hormone synthesis requires the presence of iodide; the sodium/iodide transporter (NIS) transports iodide from the blood into thyrocytes against its concentration gradient since the thyroid gland maintains a 20-40 times higher concentration of free iodide than that in the plasma (Figure 1-2) (Targovnik et al. 2010). Iodide is then transferred from thyrocytes into the colloid via an apical anion channel (Grasberger and Refetoff 2011). Iodide undergoes oxidation and is then covalently linked to tyrosine residues of thyroglobulin, this process is called iodine organification (Stathatos 2012), resulting in 3monoiodotyrosine (MIT) and 3,5-diiodotyrosine (DIT) which bind to form the hormones T3 and T4; these steps are catalyzed by the enzyme thyroid peroxidase (TPO) (Figure 1-2) (Targovnik et al. 2010; Cangul et al. 2012a). The resulting thyroglobulin containing MIT, DIT, T3 and T4, called the mature thyroglobulin, is stored in the colloid and undergoes 4 proteolysis in the lysosome to release predominantly T4 and a small amount of T3 (Figure 1-2) (Targovnik et al. 2010; Grasberger and Refetoff 2011). The released MIT and DIT undergo deiodination to release iodide which is recycled and used in further hormone synthesis (Targovnik et al. 2010; Grasberger and Refetoff 2011). Since the potent T3 is only secreted in small amounts form the thyroid, it is predominantly produced in peripheral tissues by deiodination of T4 catalyzed by one of three identified enzymes; Type-1, Type-2, and Type-3 iodothyronine deiodinases (Targovnik et al. 2010). The production of thyroid hormones is regulated by three major parts: the thyroid follicle in the thyroid gland, the hypothalamus and the pituitary gland through the action of the hypothalamic-pituitary-thyroid axis (Stathatos 2012). Thyroid Follicles Thyroid follicles are the functional units of the thyroid gland, each follicle is a cystic structure consisting of a single layer of epithelial cells, termed thyrocytes or follicular cells, that make up the wall structure of the follicle (Stathatos 2012). Each follicle is filled with the colloid, a thick sticky matter mostly consisting of thyroglobulin, which is a large glycoprotein that stores thyroid hormone (Kirsten 2000; Stathatos 2012). The thyroid gland produces thyroid hormones and stores considerable amounts of them in the colloid’s thyroglobulin until they are needed by the body (Kirsten 2000). Hypothalamic-Pituitary-Thyroid Axis The hypothalamic-pituitary-thyroid (HPT) axis regulates various functions both during development as well as in adult life through the regulation of thyroid function (Costa-e-Sousa and Hollenberg 2012; Schmaltz 2012). The hypothalamus secretes thyrotropin-releasing hormone (TRH) which in turn stimulates the anterior pituitary gland to secrete thyroid-stimulating hormone (TSH), which stimulates thyroid hormone production and is essential for thyroid function regulation (Figure 1-3) (Stathatos 2012). The HPT axis is mainly regulated by a negative feedback system between its three 5 components to ensure equilibrium of serum thyroid hormone levels (Schmaltz 2012). Small changes in serum thyroid hormone levels affect the HPT axis; high serum thyroid hormone levels directly inhibit TRH production, whereas low levels stimulate increased production of TRH in the hypothalamus leading to stimulation of TSH synthesis and increased thyroid hormone production (Figure 1-3) (Stathatos 2012). Since TSH is extremely sensitive to serum thyroid hormone levels, it is the test of choice for clinicians to diagnose most cases of thyroid dysfunction (Stathatos 2012). Thyroid Stimulating Hormone A very important regulator of thyroid function is TSH which stimulates the thyroid gland resulting in secretion of thyroid hormones (Stathatos 2012). TSH effect is exerted through a specific receptor, the TSH receptor (TSHR), found on the thyrocyte membrane (Stathatos 2012). TSHR activation results in intracellular effects mediated by adenylyl cyclase stimulation leading to increased levels of the intracellular secondary messenger, cyclic adenosine monophosphate (cAMP), which eventually results in modulation of gene expression in the thyrocytes (Szkudlinski et al. 2002; Stathatos 2012). This ultimately results in an increase in iodine uptake and organification, increased hormone production and release, as well as promotion of thyroid growth (Szkudlinski et al. 2002). Embryological Development of the Thyroid Gland Thyroid gland embryologic development starts at human embryonic day 22, with the appearance of the thyroid placode, originating from the embryonic endoderm layer (Stathatos 2012). The thyroid placode is considered the first morphological evidence for the thyroid, and is formed by a thickening of the endodermal layer in an area overarching the aortic sac (De Felice and Di Lauro 2011). However, recent molecular evidence suggests that 12 to 24 hours before the thickening occurs, specific transcription factors that are critical for thyroid development, such as NK2 homeobox 1 (NKX2-1) and paired 6 box 8 (PAX8), are expressed in a group of single layered endodermal area cells leading to their differentiation (De Felice and Di Lauro 2011). This differentiation through a specific gene expression program is considered the first event in thyroid development (De Felice and Di Lauro 2011). It is hypothesized that this monolayer of committed cells recruits other cells to form the multilayer thyroid placode (De Felice and Di Lauro 2011). The formed thyroid placode then extends into the underlying mesenchyme and migrates in a dorsal and posterior direction to form the thyroid bud (De Felice and Di Lauro 2011). The primitive thyroid gland continues to expand to form the two lobes of the thyroid (Stathatos 2012). When the thyroid cells reach their final destination anterior to the trachea, precursors of thyroid follicular cells are organized into follicles with activated terminal differentiation program leading to the expression of specific genes that devote the cells to the synthesis of thyroid hormones (De Felice and Di Lauro 2011). This process of thyroid morphogenesis is typically finalized after 49 days when the bilobed thyroid begins to form follicles and is visible anterior to the trachea (Kratzsch and Pulzer 2008). The fetal thyroid steadily increases in volume in the further course of gestation, and is capable of concentrating iodide and synthesizing thyroid hormone at about 11 to 13 weeks of gestation (Kratzsch and Pulzer 2008; Stathatos 2012). This specific sequence of embryologic events is necessary for the normal development of the thyroid gland, and any defect in these events can lead to developmental thyroid anomalies that result in disease such as CH (Stathatos 2012). Molecular Aspects of Thyroid Morphogenesis Thyroid gland morphogenesis and development depends on numerous genes (De Felice and Di Lauro 2011). During this process, regulating mechanisms can be divided into two groups; cell-autonomous and mesoderm-derived (Fagman and Nilsson 2011). Cell-autonomous regulation is activated through the transcription factors NKX2-1, FOXE1, PAX8, and HHEX co-expressed by the endoderm layer progenitor cells that are 7 committed to forming the thyroid placode (Fagman and Nilsson 2010; Fagman and Nilsson 2011). Mesoderm-derived regulation is mediated by mesoderm cells, surrounding the developing thyroid, expressing the transcription factor TBX1, which is not expressed in thyroid progenitor cells, as well as mesoderm-derived fibroblast growth factors which are essential for correct thyroid development (De Felice and Di Lauro 2011; Fagman and Nilsson 2011). Thyroid Function in the Fetus Thyroid hormones are crucial for fetal neurodevelopment (Shields et al. 2011), and the fetus relies on maternal thyroid hormones, especially during the early stages of pregnancy (Julvez et al. 2013). During development and up until the second trimester, the primitive thyroid gland is mainly nonfunctional, and fetal neurodevelopment is solely dependent on maternal thyroid hormones that enter the fetal circulation across the placenta (Schmaltz 2012). After the 12th week of gestation and until delivery, fetal TSH and T4 levels increase, whereas T3 levels remain relatively low (Kratzsch and Pulzer 2008). As thyroid hormone production from the fetus is increasing, the placenta starts to restrict the permeability of maternal thyroid hormones (Schmaltz 2012). However, there is evidence that in fetuses with CH, substantial transfer of maternal T4 across the placenta takes place even during late gestation (Vulsma et al. 1989; Shields et al. 2011). Thyroid Function in the Neonate Full Term Newborns Following delivery, the neonate undergoes a series of changes to adapt to the extrauterine environment including changes in thyroid function (Kirsten 2000; Schmaltz 2012). The dramatic decrease of temperature that the newborn experiences at delivery, results in a surge of TRH and TSH (Schmaltz 2012). TSH levels rise in the first 30 minutes after delivery and, in turn, induce increased production of T4 and T3 in the first 8 36 to 48 hours of life (Kirsten 2000). The cold-induced postnatal TSH surge does not last long and TSH levels start to decrease due to the feedback inhibition by T4, and within the second day of life, TSH levels return to normal (Kratzsch and Pulzer 2008; Schmaltz 2012). The surge in thyroid hormones T4 and T3 fades by six weeks of life (Schmaltz 2012). Preterm Newborns The hypothalamic-pituitary-thyroid (HPT) axis of preterm newborns is immature and the degree of immaturity depends on gestational age (Kratzsch and Pulzer 2008). Consequently, the cold-induced postnatal TSH surge is blunted in preterm newborns and thyroid hormone levels are decreased (Kirsten 2000). These changes in thyroid function vary between preterm newborns depending on gestational age, and thyroid hormone levels usually reach levels seen in term newborns by 38 to 42 weeks post-conceptional age (Kirsten 2000). Disorders of Thyroid Function Transient Hypothyroxinemia of Prematurity In the preterm infant, thyroid function undergoes postnatal changes related to an immature hypothalamic-pituitary-thyroid (HPT) axis, along with the interrupted exposure to maternal thyroid hormone and thyroid releasing hormone from the placenta (LaFranchi 1999). Due to immature HPT function T4 is lower in preterm infants compared to neonates born at term, and there is a direct correlation between the serum T4 level and the degree of prematurity (LaFranchi 1999; Kratzsch and Pulzer 2008). Preterm neonates with abnormal thyroid function may have transient hypothyroxinemia of prematurity (THOP), hence, it is essential to take gestational age and birth weight into consideration when making the differentiation between THOP and true cases of thyroid disorders such as congenital hypothyroidism (CH) (Hinton et al. 2010). THOP affects 35-50% of 9 preterm newborns (Berbel et al. 2010) and is characterized by low T4 levels and low to normal TSH levels (Schmaltz 2012). THOP is strongly correlated with adverse neurodevelopmental outcomes (La Gamma and Paneth 2012). However, thyroid hormone replacement therapy of newborns with THOP is controversial and is still under investigation (Kratzsch and Pulzer 2008; La Gamma and Paneth 2012; Schmaltz 2012). Congenital Hypothyroidism Congenital hypothyroidism (CH) is defined as deficient thyroid hormone production since birth (Abduljabbar and Afifi 2012). CH is a common and preventable cause of mental retardation with an incidence rate of approximately 1 in 2,350 live births (Hinton et al. 2010). Early treatment with T4 with subsequent supplementation for life produces excellent results for both growth and development (Rose et al. 2006; Hinton et al. 2010). The timing of diagnosis is crucial; the later the treatment is started, the worse the neurodevelopmental outcome and IQ will be (Kaye et al. 2006b). Infants with CH appear to be protected during the first few weeks of life due to the maternal thyroid hormone that crosses the placenta to the fetus (Kaye et al. 2006b; Abduljabbar and Afifi 2012), the best outcome is when treatment is started by two weeks of age (Rose et al. 2006). If treatment is not started, or significantly delayed, clinical features include facial puffiness, large tongue, hoarse cry, hypothermia, hypotonia, skin mottling, prolonged jaundice, poor feeding, constipation, lethargy, large fontanels, abdominal distention, and umbilical hernia (Figure 1-4) (Kaye et al. 2006b; Abduljabbar and Afifi 2012; Schmaltz 2012). CH incidence is different between ethnicities; it is higher in Hispanics, Asians, and Native Americans and lower in blacks (Hinton et al. 2010; Abduljabbar and Afifi 2012). The variable incidence rates in different ethnicities suggests the involvement of genetic factors that have not been discovered yet (Rastogi and LaFranchi 2010). CH incidence is also affected by gender where it is twice as common in females compared to 10 males (Rose et al. 2006; Hinton et al. 2010). The cause for the increased risk in females is not well known but is thought to be related to autoimmune risk, which is generally more common in females (Kaye et al. 2006b; Rastogi and LaFranchi 2010). Newborns with Down syndrome also have increased CH risk at approximately 1 in 140 (Kaye et al. 2006b). Newborn Screening CH is detected shortly after birth through newborn screening. Newborn screening is a critical public health program started in the 1960s (Wilcken and Wiley 2008). Newborn screening programs are designed to detect inborn disorders that, if left untreated, can lead to early mortality or lifelong morbidity and disability (Center for Disease Control 2001; Watson et al. 2006). It is mandatory to offer newborn screening to all infants born in the United States, however, parents have the right to opt-out of the screening (Kaye et al. 2006a; Watson et al. 2006). Screening is done shortly after birth, usually within the first 48 hours of life (Watson et al. 2006), using blood spotted on a special card through a heel prick (Kaye et al. 2006a). The timing of newborn screening sampling is important since most screened analytes have normal ranges that vary considerably within the first hours or days of life (Wilcken and Wiley 2008). For example, the TSH surge that occurs immediately after birth may result in a high rate of false positive results for CH screening when sampling is done before 48 hours of life (Kaye et al. 2006a). Approximately 4 million newborns are screened in the United States every year, and of those 3,000 are identified with a severe condition (Center for Disease Control 2001). Newborn screening programs vary between states in the conditions screened for and the technology used (Kaye et al. 2006a) There were no national standards for newborn screening programs until 2006, when the American College of Medical Genetics (ACMG) published a report with guidelines for state newborn screening programs after 11 being commissioned to do so by the Maternal and Child Health Bureau (Watson et al. 2006).The ACMG recommends mandated screening for 29 identified disorders (Watson et al. 2006). The disorders screened for include hereditary metabolic disorders (amino acid disorders, fatty acid oxidation disorders, and organic acid disorders), congenital endocrinopathies, hemoglobinopathies, and cystic fibrosis (Center for Disease Control 2001; Kaye et al. 2006a). Furthermore, many infants are screened for congenital hearing loss as part of the newborn screening program (Kaye et al. 2006a). The Iowa Neonatal Metabolic Screening Program (INMSP) screens for all 29 disorders recommended by the American College of Medical Genetics and an additional 14 metabolic conditions making it one of the most comprehensive newborn screening programs in the United States (National Newborn Screening and Genetics Resource Center 2013). CH screening was added to existing newborn screening programs in the mid 1970s (Kaye et al. 2006b). CH is screened for at birth in the newborn screening program through detection of T4, TSH, or both (Wilcken and Wiley 2008). There is no consensus on a single best CH screening protocol; 131 newborn screening programs worldwide use TSH for CH screening and 75 use a T4/TSH screening protocol, but all screening is done using an immunoassay technique (Pass and Neto 2009). Each screening method misses certain CH syndromes, and the best protocol is simultaneous testing of T4 and TSH at the same time, which is implemented in 5 states, but was considered expensive and time consuming until the recent development of multi-analyte assays that can test several biomarkers in the same assay well simultaneously (Pass and Neto 2009). The Iowa Neonatal Metabolic Screening Program (INMSP) uses TSH only for CH screening (Pass and Neto 2009). Unfortunately, newborn screening for congenital hypothyroidism has the highest number of false positive results with repeated testing costs of more than two million dollars annually (Kwon and Farrell 2000). It was estimated that only 3.9% of screen positive newborns are true positives who get diagnosed with CH (Pass and Neto 2009). 12 The false positive result rate is usually higher in newborn screening programs that use T4 as a primary analyte (0.30%) compared to programs that use TSH (0.05%) (Kaye et al. 2006b). However, most newborn screening programs in the United States still use T4 as the primary analyte, and an advantage of this is the ability to identify infants with secondary hypothyroidism that have no TSH surge, or infants with delayed TSH surge such as preterm infants, both of which would be missed in programs that measure TSH as the primary analyte (Kaye et al. 2006b). Preterm infants account for the largest percentage of newborns with false positive CH screening results due to their low T4 levels resulting from the immature HPT axis (Kaye et al. 2006b). All newborns with T4 level below the 10th percentile in the newborn screening programs that use T4 as an initial analyte, get a follow-up TSH screen (Kaye et al. 2006b). If final newborn screen results for CH are abnormal, the infant must be recalled immediately for follow-up confirmatory serum testing of thyroid function (Kaye et al. 2006b; Rastogi and LaFranchi 2010). Confirmatory serum testing includes TSH, free or total T4, and a binding protein measure such as T3 resin uptake, and is usually done around one to two weeks of age (Rastogi and LaFranchi 2010). Once a CH diagnosis is confirmed, treatment should be started immediately, and then additional optional diagnostic studies may be done to determine the underlying etiology of CH (Kaye et al. 2006b). Etiology of Congenital Hypothyroidism Congenital hypothyroidism (CH) is a heterogeneous disorder and is classified into two forms; transient and permanent (Abduljabbar and Afifi 2012). Transient Congenital Hypothyroidism Transient CH is a temporary thyroid hormone deficiency that is present at birth, but normal thyroid hormone production is recovered during the first months or years of life (Abduljabbar and Afifi 2012). Transient CH etiology is classified into maternal or 13 neonatal factors. Maternal factors include transplacental passage of antithyroid medications, TSH receptor antibodies, and iodine deficiency or excess (Rastogi and LaFranchi 2010). Neonatal factors include iodine deficiency or excess, heterozygous mutations in DUOX2 and DUOXA2 genes leading to transient or permanent CH, and congenital liver hemangiomas (Rastogi and LaFranchi 2010; Abduljabbar and Afifi 2012). Transient CH is more common in iodine deficient parts of the world such as Europe where the incidence is reported to be 1 in 100, whereas in the United States, an iodine sufficient country, the incidence is 1 in 50,000 (Rastogi and LaFranchi 2010). Permanent Congenital Hypothyroidism Permanent CH is, by definition, a persistent thyroid hormone deficiency that requires thyroid hormone replacement therapy for life (Abduljabbar and Afifi 2012). Permanent CH etiology is classified into primary or secondary. Primary Congenital Hypothyroidism Primary CH causes include abnormal development of the thyroid gland (thyroid dysgenesis, agenesis, or ectopy), abnormal thyroid hormone production from a structurally normal gland (thyroid dyshormonogenesis), and TSH binding or signaling defects (Rose et al. 2006; Rastogi and LaFranchi 2010). In iodine sufficient countries, abnormal thyroid gland development is the most common cause of CH accounting for 8085% of CH cases (Hinton et al. 2010; Rastogi and LaFranchi 2010). Thyroid dysgenesis CH is mostly sporadic; only 2% of cases have a known causing mutation in one of the genes necessary for thyroid development (NKX2.1, NKX2.5, PAX8, or FOXE1), whereas the remaining 98% of cases have an unknown cause (Rastogi and LaFranchi 2010; Abduljabbar and Afifi 2012). In the cases with a mutation in one of the genes encoding a transcription factor, CH may be present as part of a syndrome; for example FOXE1 mutations lead to Bamforth-Lazarus Syndrome (Rastogi and LaFranchi 2010). 14 The remaining ~15% of CH cases have a molecular defect in one of the genes in the thyroid hormone biosynthesis pathway most commonly inherited as autosomal recessives (Abduljabbar and Afifi 2012). Most commonly the defect is in a gene affecting the activity of the enzyme thyroid peroxidase such as DUOX2, DUOXA2, and SLC26A4. Rarely, cases of dyshormonogenesis result from a mutation in the gene encoding the sodium/iodide transporter (SLC5A5) (Rastogi and LaFranchi 2010). Secondary or Central Congenital Hypothyroidism Secondary CH, also called central CH, generally results from TSH production defects, and is most commonly part of congenital hypopituitarism (Rastogi and LaFranchi 2010). In rare cases, certain genetic mutations result in secondary CH; these include mutations in the thyrotropin releasing hormone (TRH) receptor gene leading to TRH resistance, and mutations in the gene encoding the TSH β subunit leading to TSH deficiency(Rastogi and LaFranchi 2010). Adult-Onset Hypothyroidism Adult-onset hypothyroidism results from inadequate thyroid hormone production or action. It is a common disorder affecting about 0.3% of the population in the United States, and it is more prevalent in women, the elderly, and certain ethnic groups. Just like congenital hypothyroidism (CH), adult-onset hypothyroidism may be transient or permanent, and can be primary, resulting from thyroid gland abnormalities, or secondary, resulting from abnormalities in the hypothalamic-pituitary-thyroid axis (Almandoz and Gharib 2012). The most common cause of hypothyroidism worldwide is iodine deficiency, however, in iodine sufficient areas, autoimmune disease, known as Hashimoto’s thyroiditis, is the leading cause (Vanderpump 2011). Hypothyroidism symptoms are often subtle but may include fatigue, muscle aches, constipation, dry skin, cold intolerance, and vocal changes (Almandoz and Gharib 2012). If left untreated, hypothyroidism can affect blood pressure, lipid levels, fertility, cognition, and 15 neuromuscular function (Gaitonde et al. 2012). The best laboratory test for hypothyroidism is serum thyroid stimulating hormone (TSH) measurement, if it is found to be elevated, it is followed by a serum free thyroxine (FT4) measurement. Hypothyroidism is diagnosed based on elevated serum TSH levels with low serum FT4 levels, and is treated with thyroid hormone replacement therapy (Almandoz and Gharib 2012; Gaitonde et al. 2012). Genetics of Thyroid Stimulating Hormone The hypothalamic-pituitary-thyroid axis is complexly regulated both by genetic as well as physiological factors, leading to the individuality of thyroid hormone and TSH levels in each subject both in normality and in thyroid dysfunctional states (Midgley et al. 2013). This is observed in the distribution of TSH levels in the population versus in an individual; it was found that TSH has a wide population reference range whereas the range is much narrower in the same individual TSH levels over a period of time (Andersen et al. 2002; Panicker 2011; Hoermann and Midgley 2012; Midgley et al. 2013). Several studies tried to estimate the genetic contribution to this individuality in TSH levels through twin-based or family-based study designs (Panicker 2011). TSH heritability was estimated to vary from 32% (Samollow et al. 2004), to 64% (Hansen et al. 2004), and to a maximum of 65% (Panicker et al. 2008). Heritability is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic factors. The heritability of a given phenotype can be estimated from the difference of trait variation between monozygotic and dizygotic twin pairs (Boomsma et al. 2002). There have been several genome-wide association studies (GWAS) as well as candidate-gene studies to identify the genes that are responsible for the heritability of TSH levels and thyroid function. 16 Genome-Wide Association Studies Genome-wide association studies (GWAS) are designed to search for common genetic variants that are associated with complex traits or diseases through the genotyping of hundreds of thousands to millions of single nucleotide polymorphisms (SNPs) across the genome in hundreds or even thousands of individuals (Manolio 2010). GWAS allow for interrogating the entire genome without a prior hypothesis or premeditated candidate gene selection (Dube and Hegele 2013). SNPs that are found to be associated with a trait or disease in GWAS are typically not the causative variant, but are surrogates for most other variants, and potentially the causative variant, in nearby loci through linkage disequilibrium (LD) (Dube and Hegele 2013). LD implies that SNPs are transmitted from one generation to the next in blocks, and genotyping a few SNPs in each block, called tag SNPs, can capture the variation of most other SNPs within that block (Manolio 2010). This LD phenomenon is the result of the low recombination rate of approximately 1 crossover per 100 megabases per generation (Altshuler et al. 2008). However, the transition from a GWAS significantly associated SNP to the identification of the specific etiologic variant that is tagged by the GWAS signal is still challenging (Dixon et al. 2011). To date, six GWAS and two meta-analyses have been published with TSH level as one of the phenotypes studied (Table 1-1) (Hwang et al. 2007; Arnaud-Lopez et al. 2008; Gudmundsson et al. 2009; Lowe et al. 2009; Panicker et al. 2010; Gudmundsson et al. 2012; Rawal et al. 2012; Porcu et al. 2013). The GWAS by Hwang et al. (2007) studied 810 individuals form the Framingham Heart Study for genetic association with mean TSH levels from two measurements. Two of the top three significantly associated SNPs are intergenic on chromosome 7 and the third SNP is in an intron of HACE1 (Hwang et al. 2007). The second TSH related GWAS was done by Arnaud-Lopez et al. (2008) on 4,300 Sardinians, and the strongest identified association was for an intronic SNP in 17 PDE8B (Figure 1-5). This association was successfully replicated in an additional 4,158 individuals form Sardinia, Tuscany, and the Old Order Amish (Arnaud-Lopez et al. 2008). The PDE8B gene encodes a high affinity adenosine 3’,5’-cyclic monophosphate (cAMP)-specific phosphodiesterase to regulate the level of cAMP in cells and plays a vital role in signal transduction (Hayashi et al. 1998; Lakics et al. 2010). In the ArnaudLopez et al. paper, the authors suggest that since cAMP is necessary for thyroid-hormone secretion due to TSH stimulation; when PDE8B catalyzes the hydrolysis and inactivation of cAMP in the thyroid gland, it results in decreased generation of thyroid-hormone T4 and T3 resulting in the negative feedback loop to act on producing more TSH (ArnaudLopez et al. 2008). The third GWAS done by Lowe et al. (2009) on over 2,800 Micronesian individuals found a novel significant association for 10 SNPs in a region on chromosome 9 encompassing the gene encoding thyroid transcription factor 2 (TTF-2, now known as FOXE1) with TSH levels. This was the first report of an association for this locus with the variation of TSH levels, which is interestingly, a strong biological candidate gene (Lowe et al. 2009). FOXE1 gene encodes a transcription factor that is essential for the initiation of thyroid differentiation at the embryonic stage (Parlato et al. 2004). Mutations of the FOXE1 gene may result in thyroid dysgenesis leading to congenital hypothyroidism (Castanet and Polak 2010). FOXE1 also plays an important role in regulating the transcription of different thyroid-specific genes resulting in regulation of thyroid-hormone synthesis (Gudmundsson et al. 2009). This GWAS also replicated the PDE8B finding reported previously in Caucasians by Arnaud-Lopez et al. (2008). Next, Gudmundsson et al. (2009) studied the genetic association with serum TSH levels in a population of ~12,000 Icelanders for two SNPs that were previously found to be significantly associated with thyroid cancer in a GWAS. The two intergenic SNPs were found to be significantly associated with serum TSH levels; one is on chromosome 9 and the other is on chromosome 14, with FOXE1 and NKX2-1 being the nearest genes 18 respectively (Gudmundsson et al. 2009). NKX2-1 and FOXE1 are plausible candidates given their involvement in thyroid gland development; both genes encode transcription factors necessary for proper thyroid organogenesis and are referred to as thyroid transcription factor 1 and 2 (TTF1 and TTF2) respectively (Gudmundsson et al. 2009). The fifth GWAS done by Panicker et al. (2010) on 2,014 twins from the United Kingdom identified a novel locus on chromosome 1, near CAPZB gene, to be significantly associated with serum TSH levels (Figure 1-6). This finding was successfully replicated in an independent cohort of 1,093 Australian individuals (Panicker et al. 2010). CAPZB gene encodes the beta subunit of an actin binding protein that helps regulate the growth of the actin filament. It is known to be expressed in the thyroid, brain, and pituitary, making it a plausible candidate for involvement in pituitarythyroid axis regulation, however, the mechanism is not known yet (Panicker et al. 2010). The sixth GWAS done by Gudmundsson et al. (2012) on 27,758 Icelandic individuals reported 22 SNPs to be significantly associated with serum TSH levels at a pvalue <5x10-8. Of the loci previously identified to be associated with TSH levels; the PDE8B locus (Arnaud-Lopez et al. 2008), the chromosome 9 and chromosome 14 loci (Gudmundsson et al. 2009), and the chromosome 1 locus (Panicker et al. 2010), were replicated in this GWAS (Gudmundsson et al. 2012). Rawal et al. (2012) performed a meta-analysis of two GWAS, with a total sample size of 3736 individuals of European ancestry, to identify genetic associations with TSH levels. The meta-analysis results showed statistically significant associations with four loci (Figure 1-7), two of which replicate previous GWAS findings for PDE8B on chromosome 5 (Arnaud-Lopez et al. 2008), and CAPZB on chromosome 1 (Panicker et al. 2010). The third locus is in an intergenic region on chromosome 16, near MAF, that region was previously found in a GWAS to be associated with thyroid volume and goiter (Teumer et al. 2011). The forth locus is on chromosome 4 upstream of NR3C2, and this is the first report of an association between this locus and TSH levels (Rawal et al. 2012). 19 NR3C2 encodes the mineralocorticoid receptor that mediates the actions of aldosterone and functions as a transcription factor. It is highly expressed in the thyroid gland, and the authors hypothesize that it may be involved in regulating the expression of thyroid specific genes involved in the transduction of the received TSH/cAMP signal (Rawal et al. 2012). All four loci were successfully replicated in 3,727 individuals of European ancestry (Rawal et al. 2012). Recently, Porcu et al. (2013) performed a large meta-analysis of GWAS to identify genetic variants associated with serum TSH in over 26,000 euthyroid European individuals from 18 studies. The authors reported 19 loci to be significantly associated with TSH levels (Figure 1-8). Four loci replicate previous GWAS findings for PDE8B (Arnaud-Lopez et al. 2008), CAPZB (Panicker et al. 2010), NR3C2 and chromosome 16 region, near MAF (Rawal et al. 2012), whereas the remaining 15 loci are novel (Porcu et al. 2013). Candidate-Gene Studies Candidate-gene studies select genes that are suspected to play a role in thyroid physiology based on biological information, the selected genes are then studied and fine mapped for associations (Panicker 2011). Candidate-gene studies identified a polymorphism in the TSH receptor gene (TSHR) to be associated with adult serum TSH levels (Hansen et al. 2007). However, this finding was not replicated in GWAS (Panicker 2011). The lack of reproducibility for candidate-gene studies in GWAS is a common occurrence in complex traits and diseases. A possible explanation for this is publication bias, which refers to the higher likelihood of publication for positive results compared to negative results, implying the possibility that those were not true associations, but rather false positive findings (Bosker et al. 2011). However, other explanations are that phenotyping of complex traits and diseases across different studies may vary, and that complex traits and diseases are affected by gene-gene and gene-environment interactions 20 that may be different across populations, and all this variability may lead to the lack of reproducibility (Bosker et al. 2011). Therefore, it has been argued that GWAS, at this point, are not designed to definitively exclude the role of possible candidate genes in complex traits or diseases (Michel et al. 2010). Many other biologically plausible genes, such as the genes encoding thyroid hormone receptors (THRA and THRB) and thyroid hormone transporter (MCT8) have been studied in candidate-gene studies but have not shown significant associations (Panicker 2011). The three genes encoding the three iodothyronine deiodinases (DIO1, DIO2, and DIO3) have also been studied because of their vital role in thyroid function, but no association with serum TSH levels was found (Panicker 2011). Main Thyroid Related Genes PDE8B The phosphodiesterase 8B (PDE8B) gene is located on chromosome 5. It is expressed most abundantly in the thyroid gland where it has threefold higher level than in the next highest tissue (Lakics et al. 2010). It is expressed at lower levels in some other tissues including the brain, spinal cord, and placenta (Hayashi et al. 1998). The PDE8B gene encodes a high affinity adenosine 3’,5’-cyclic monophosphate (cAMP)-specific phosphodiesterase that catalyzes hydrolysis of cAMP to regulate its level in cells, and therefore plays a vital role in signal transduction (Hayashi et al. 1998; Lakics et al. 2010; Panicker 2011). Mutations of PDE8B result in the autosomal-dominant striatal degeneration (ADSD) disease (Appenzeller et al. 2010). However, a study of a large family with ADSD did not find any thyroid function abnormalities in affected individuals (Kuhlenbaumer et al. 2004). PDE8B mutations were also found in patients with adrenal hyperplasia and Cushing syndrome (Horvath et al. 2008). 21 NKX2-1 NK2 homeobox 1 (NKX2-1) gene, also called thyroid transcription factor 1 (TTF1), encodes a transcription factor that is necessary for the regulation of thyroid specific gene expression as well as thyroid morphogenesis (Parlato et al. 2004). Mutations in this gene are associated with congenital hypothyroidism due to thyroid dysgenesis (Rastogi and LaFranchi 2010). A role of NKX2-1 in regulating TSH level is supported by two GWAS (Gudmundsson et al. 2009; Gudmundsson et al. 2012). FOXE1 Forkhead box E1 (FOXE1) gene, also called thyroid transcription factor 2 (TTF2), is located on chromosome 9. FOXE1 encodes a transcription factor that is essential for the initiation of thyroid differentiation at the embryonic stage (Parlato et al. 2004). Mutations of the FOXE1 gene may result in thyroid dysgenesis leading to both familial as well as cases of syndromic congenital hypothyroidism in the Bamforth-Lazarus Syndrome, Online Mendelian Inheritance in Man (OMIM) #241850 (Online Mendelian Inheritance in Man 2013), a rare inherited disorder characterized by CH, cleft palate, and spiky hair (Castanet and Polak 2010). FOXE1 also plays an important role in regulating the transcription of different thyroid-specific genes resulting in regulation of thyroidhormone synthesis (Gudmundsson et al. 2009). TSHR Thyroid stimulating hormone receptor (TSHR) gene is located on chromosome 14. The protein encoded by the TSHR gene is the TSH receptor (TSHR). TSHR is present on thyroid cells, and when activated by TSH secreted from the pituitary gland, intracellular cAMP is upregulated resulting in activation of various cellular processes ending with an increased production of thyroid hormone (Stathatos 2012). TSHR activating mutations cause several types of hyperthyroidism (Hebrant et al. 2011). TSHR loss of function 22 mutations may lead to an autosomal recessive form of thyroid dysgenesis (Cangul et al. 2012b). CAPZB F-actin-capping protein subunit beta (CAPZB) gene encodes the beta subunit of an actin binding protein that helps regulate the growth of the actin filament. It was found to be associated with TSH levels in two GWAS and two meta-analyses (Panicker et al. 2010; Gudmundsson et al. 2012; Rawal et al. 2012; Porcu et al. 2013). It is known to be expressed in the thyroid, brain, and pituitary, making it a plausible candidate for involvement in pituitary-thyroid axis regulation, however, the mechanism is not known yet (Panicker et al. 2010; Panicker 2011). Purpose of Study It is well established that genetic variants play an important role in defining thyroid function throughout life, both in health as well as in disease states. The role of genetics has been extensively studied through GWAS and candidate-gene studies, and several genes have been identified to be associated with the variation of thyroid function between individuals. Elucidating the genetic basis of thyroid function variability is currently an area of interest not only to advance our understanding of thyroid physiology, but also to further the understanding of adult conditions related to thyroid function. For example, low serum TSH levels are associated with an increased risk of atrial fibrillation in adults over 60 years old (Sawin et al. 1994). Thyroid hormone levels within the normal physiological range have also been shown to affect bone mass and density in healthy men aged 25-45 years (Roef et al. 2011), as well as in men and women above 55 years of age (van der Deure et al. 2008). Furthermore, some of the genetic variation reported to be associated with thyroid function was also found to be associated with several other clinical and developmental phenotypes (Panicker 2011). For example, variants in DIO2 23 were found to be associated with osteoarthritis, and variants in TSHR were found to be associated with bone density, insulin resistance, and longevity (Panicker 2011). Although there have been many significant new findings in the genetics of thyroid function over the past decade, we are still at the beginning and there are more genes and pathways yet to be discovered (Panicker 2011). Furthermore, most studies address thyroid function in adulthood and none in the neonatal phase. The purpose of this study is to examine genetic associations with thyroid function, namely TSH level variation, in the neonatal period. This will enhance our understanding of thyroid function in newborns, as well as potentially identify avenues for the early prediction of adult diseases that are associated with thyroid function genetic variants. Hypothesis Genetic polymorphisms within biologically relevant pathways, including neuroendocrine pathways, are associated with the normal variation of TSH levels in neonates. Specific Aims Specific Aim 1: Estimate the heritability of TSH level using newborn blood spot cards of monozygotic and dizygotic twin pairs. Specific Aim 2: Identify SNPs associated with normal variation in TSH measurements, obtained from the Iowa Neonatal Metabolic Screening Program, in euthyroid preterm and term infants. Specific Aim 3: Fine mapping of potential FOXE1 and PDE8B enhancer regions to identify genetic variants that are associated with normal variation in neonatal TSH measurements. 24 Table 1-1 Summary of GWAS data for TSH GWAS Associated Locus Candidate Gene in Region P-Value Hwang et al. 2007 7p21 6q16 7p15 Intergenic HACE1 Intergenic 4 x 10-6 7 x 10-6 8 x 10-6 Arnaud-Lopez et al. 2008 5q13 PDE8B 1 x 10-11 Lowe et al. 2009 9q22 5q13 FOXE1 PDE8B 1 x 10-6 3 x 10-4 Gudmundsson et al. 2009 9q22 14q13 FOXE1 NKX2-1 3 x 10-14 3 x 10-2 Panicker et al. 2010 1p36 CAPZB 3 x 10-8 Gudmundsson et al. 2012 5q13 9q22 1p36 14q13 PDE8B FOXE1 CAPZB NKX2-1 3 x 10-62 4 x 10-31 4 x 10-26 1 x 10-16 Rawal et al. 2012 5q13 4q31 16q23 1p36 PDE8B NR3C2 MAF CAPZB 3 x 10-27 3 x 10-10 6 x 10-10 2 x 10-8 Porcu et al. 2013 5q13 1p36 16q23 4q31 PDE8B CAPZB MAF NR3C2 2 x 10-56 4 x 10-21 9 x 10-18 9 x 10-16 25 Figure 1-1 Thyroid gland. Anterior view of the thyroid gland showing its location below the larynx and anterior to the trachea. Source: (Kirsten 2000). Reprinted with permission from Springer Publishing Company: Neonatal Network, Volume 19, Number 8, Endocrine Series #6, Figure 1. 26 Figure 1-2 Iodine organification and thyroid hormone synthesis. Source: (Stathatos 2012). Reprinted with permission from Elsevier. 27 Figure 1-3 Hypothalamic-pituitary-thyroid axis regulation. Source: (Stathatos 2012). Reprinted with permission from Elsevier. 28 Figure 1-4 Three month old infant with untreated congenital hypothyroidism. (A) This picture demonstrates hypotonia. (B) This picture demonstrates facial puffiness, large tongue, large fontanels, and skin mottling. (C) This picture demonstrates abdominal distention, and umbilical hernia. Source: (Rastogi and LaFranchi 2010). Reprinted with permission from BioMed Central. 29 Figure 1-5 A Manhattan plot of the TSH level GWAS done by (Arnaud-Lopez et al. 2008). SNPs are arranged by chromosome number and plotted on the X-axis. The Y-axis is the –log10 of the association p-value. Source: (Arnaud-Lopez et al. 2008). Reprinted with permission from Elsevier. 30 Figure 1-6 A Manhattan plot of the TSH level GWAS done by (Panicker et al. 2010). SNPs are arranged by chromosome number and plotted on the X-axis. The Y-axis is the – log10 of the association p-value. Source: modified from (Panicker et al. 2010). Reprinted with permission from Elsevier. 31 Figure 1-7 A Manhattan plot of the TSH level GWAS meta-analysis done by (Rawal et al. 2012). SNPs are arranged by chromosome number and plotted on the X-axis. The Yaxis is the –log10 of the association p-value. The upper horizontal line indicates the genome-wide threshold of statistical significance of p-value of 5 x 10-8. Source: (Rawal et al. 2012). Reprinted with permission from Oxford University Press. 32 Figure 1-8 A Manhattan plot of the TSH level GWAS meta-analysis done by (Porcu et al. 2013). SNPs are arranged by chromosome number and plotted on the X-axis. The Y-axis is the –log10 of the association p-value. The horizontal line indicates the genome-wide threshold of statistical significance of p-value of 5 x 10-8. The loci that reached genomewide significance are highlighted in green. Source: Modified from (Porcu et al. 2013). Reprinted form PLOS Genetics, no permission required under the Creative Commons Attribution License. 33 CHAPTER 2 THE HERITABILITY OF METABOLIC PROFILES IN NEWBORN TWINS The following manuscript was published in Heredity, 2013; 110: 253-258. Additional authors include Daniel Cook, Oleg Shchelochkov, Lauren Fleener, Stanton Berberich, Jeffrey Murray, and Kelli Ryckman. The objective of the work described in this chapter is to estimate the heritability of thyroid stimulating hormone (TSH) and other metabolites routinely measured in newborns. TSH heritability of up to 65% has been estimated in adults; however, to-date has not been reported in neonates. Our study is the first to assess heritability of TSH levels in the neonatal period, as well as the first to use newborn blood spot card measurements to estimate the genetic heritability of any trait, illustrating the utility of stored newborn blood spot cards as a valuable resource for genetic studies. We estimated the heritability of neonatal TSH at 58% which is consistent with the previous reports of TSH heritability in adults. This study provides direct evidence for a strong genetic contribution to TSH variability at birth. Drs. Jeffrey Murray and Kelli Ryckman provided oversight for all aspects of the project, provided input into study design and assisted in the writing and editing of the manuscript. Dr. Ryckman also performed statistical analysis with the assistance of Daniel Cook. Dr. Oleg Shchelochkov provided valuable insight into newborn screening and editing the manuscript. Lauren Fleener performed TaqMan genotyping. Dr. Stanton Berberich provided input into the study design and access to study samples and data. I was involved in study design, literature review, performed DNA extraction from newborn blood spot cards for all twin samples and assisted in the analysis. I also wrote the manuscript, revisions, and responses to reviewer critiques. 34 Abstract Identifying genetic and metabolic biomarkers in neonates has the potential to improve diagnosis and treatment of common complex neonatal diseases, and potentially lead to risk assessment and preventative measures for common adulthood illnesses such as diabetes and cardiovascular disease. There is a wealth of information on using fatty acid, amino acid, and organic acid metabolite profiles to identify rare inherited congenital diseases through newborn screening, but little is known about these metabolic profiles in the context of the “healthy” newborn. Recent studies have implicated many of the amino acid and fatty acid metabolites utilized in newborn screening in common complex adult diseases such as cardiovascular disease, insulin resistance, and obesity. To determine the heritability of metabolic profiles in newborns, we examined 381 twin pairs obtained from the Iowa Neonatal Metabolic Screening Program. Heritability was estimated using multilevel mixed-effects linear regression adjusting for gestational age, gender, weight and age at time of sample collection. The highest heritability was for short chain acylcarnitines, specifically butyrylcarnitine (C4) (h2=0.66, P=2x10-16), methylmalonylcarnitine (C4-DC) (h2=0.83, P<10-16) and isovalerylcarnitine (C5) (h2=0.61, P=1x10-9). Thyroid stimulating hormone (TSH) (h2=0.58, P=2x10-5) and immunoreactive trypsinogen (IRT) (h2=0.52, P=3x10-9) also have a strong genetic component. This is direct evidence for a strong genetic contribution to the metabolic profile at birth and that newborn screening data can be utilized for studying the genetic regulation of many clinically relevant metabolites. Introduction Metabolomics is the study of metabolites, small molecules that make up the building blocks of cellular processes (Goonewardena et al. 2010). Metabolomics is a rapidly developing field of great importance in understanding the physiology of dynamic cellular processes and for diagnosis of human diseases. Better understanding of the 35 metabolic profiles present in the immediate postnatal period is important for their short term contribution to diagnosis as well as for the long term prediction of outcomes. Metabolic profiles of the neonate, defined by levels of amino acid, organic acid, and fatty acid oxidation metabolites, may be correlated with metabolic conditions in adulthood, such as obesity, diabetes and cardiovascular disease through the phenomenon of metabolic programming (Srinivasan and Patel 2008). Understanding the genetic contribution to metabolic profiles, particularly in the neonate, may provide insight into complex diseases and their risks. The underlying genetic contribution to metabolic traits has been studied in plants (Keurentjes et al. 2006), mice (Ferrara et al. 2008), and more recently, in adult humans (Shah et al. 2009). However, to-date, we are aware of no studies examining the heritability of metabolic traits related to amino acid and fatty acid oxidation in the neonatal period. Population-based metabolic profiling is most commonly implemented in Statemandated neonatal screening programs used to detect numerous endocrine and metabolic disorders at birth. Levels of amino acids, including branched-chain amino acids, and acylcarnitines are measured to detect newborns with rare disorders that are treatable, if detected early (Wilcken and Wiley 2008). Some hormones and enzyme activities are also measured including thyroid stimulating hormone (TSH), immunoreactive trypsinogen (IRT), and 17-hydroxyprogesterone (17-OHP) to detect disorders such as congenital hypothyroidism, cystic fibrosis, and congenital adrenal hyperplasia respectively (Center for Disease Control 2001; Khoury et al. 2003; Votava et al. 2005). In addition to detecting disorders at birth, branched-chain amino acids have been associated with adult diseases including type 2 diabetes and cardiovascular disease (Shah et al. 2010; Wang et al. 2011). There are several genome-wide association studies that have identified genetic loci associated with metabolite measurements from adult human blood and/or urine samples (Gieger et al. 2008; Illig et al. 2010; Suhre et al. 2011a; Suhre et al. 2011b; 36 Kettunen et al. 2012). While initial results are promising, less effort has been devoted to identifying the underlying heritability of metabolic traits in humans at different times of development, particularly at birth. The heritability of biomarkers for amino acid and fatty acid oxidation is virtually unexplored in adults or neonates with the exception of a single study examining an extensive panel of metabolic markers from adult plasma samples (Shah et al. 2009). The authors demonstrated high heritability among several amino acids and acylcarnitines in eight multiplex families with a family history of premature cardiovascular disease (Shah et al. 2009). A recent study by Kettunen et al. examined the heritability for amino acids, lipoproteins, lipids, and some other metabolites such as glucose and urea in adult serum samples; however, the paper does not investigate acylcarnitines which are important for fatty acid oxidation (Kettunen et al. 2012). An additional study by Nicholson et al. investigates the genetic and environmental contribution to human metabolic profiles, and the authors report estimations of familial variation for different metabolite measurements; however they state that their sample size is insufficient for heritability estimations (Nicholson et al. 2011). A few other studies have shown a high degree of heritability in adult hormone measurements, particularly, adult serum levels of TSH where heritability estimates ranged from 32% (Samollow et al. 2004) to 64% (Hansen et al. 2004) to 65% (Panicker et al. 2008). In this study, we obtained twin samples from the Iowa Neonatal Metabolic Screening Program (INMSP) to determine the heritability of hormone, enzyme, acylcarnitine, and amino acid measurements measured during routine newborn screening. Understanding the heritability of these analytes at birth may increase our understanding of physiologic processes and metabolic programming in various adulthood diseases. Furthermore, identifying the heritable components of the metabolome, particularly in the neonatal period, may pave the way for future genetic association studies that may provide insight into the physiology of different cellular processes, as well as the biology of complex neonatal and adulthood diseases. 37 Methods Study Population Metabolic data for 47 analytes from routine newborn screening were obtained from the University of Iowa State Hygienic Laboratory (SHL) for 243 same-sex twin pairs (130 male-male twin pairs and 113 female-female twin pairs) and 165 male-female twin pairs born in 2009 (Table 2-1). Twins were identified by the SHL based on both infants having the same birth date, gestational age, mother’s first name and facility identification number. Newborn dried blood spots (DBS) were obtained from a heel stick 24-72 hours after birth. DBS specimens were collected, dried and handled according to the Clinical Laboratory Standards Institute (CLSI) guideline (Hannon et al. 2007). DBS specimens were transported to the SHL within 5 days of collection, and evaluated for quality at the time of arrival by trained technical staff. Blood spot cards were obtained for DNA extraction and zygosity testing for all same-sex twin pairs. Approval for use of the de-identified data and blood spot cards was granted by the Iowa Department of Public Health and a waiver of consent was obtained from the Institutional Review Board of the University of Iowa (IRB#200908793). DNA was extracted from one dried whole blood spot using the AutoGen (Holliston, MA, USA) QuickGene-810 nucleic acid extraction machine with the DNA Tissue Kit (catalogue number DT-S) and following manufacturer’s recommendations. Enzymatic Assays Quantification of 17-hydroxyprogesterone (17-OHP), TSH and IRT were determined by solid phase, time-resolved fluoroimmunoassay from dried newborn blood spots using Perkin Elmer’s AutoDELFIA® platform (Waltham, MA, USA). Galactose-1phosphate uridyl transferase (GALT) was determined by a semi-quantitative enzymatic assay by Perkin Elmer (Waltham, MA, USA) based on the Beutler method. 38 Tandem Mass Spectrometry Tandem mass spectrometry is performed in neonatal screening to detect levels of amino acids and acylcarnitines. Screening procedures in Iowa are based on previously established methodology (Chace et al. 2001; Turgeon et al. 2008; Chace et al. 2009). Briefly, a derivatization method is used in which butyl esters of acylcarnitines and amino acids are prepared from the extracts. For succinylacetone, hydrazine derivatives are prepared. Tandem mass spectrometry is performed with Waters Quattro Micro triple quadrupole tandem mass spectrometers, equipped with an electrospray ionization source operated in the positive ion mode. Sensitivity and resolution checks on the instruments are performed daily and calibration and mass accuracy adjustments are done 2 – 3 times per year. Multiple reaction monitoring (MRM) mode is used to scan for specific mass ion intensities. Concentrations are obtained from the ratio of ion intensity (cps) at the mass that represents a specific analyte compared with its isotopically labeled internal standard and correcting for blood volume in a 1/8 inch DBS punch. Both internal and external spiked control specimens, a normal control specimen, and a blank are analyzed with each batch of specimens. The external spiked control specimens are obtained from Newborn Screening Quality Assurance Program at the Centers for Disease Control. Determination of Zygosity Zygosity was determined based on concordance of genetic markers by genotyping 20 markers with high heterozygosity (~0.5) (Table 2-2) that were not in linkage disequilibrium with one another. Genotyping was performed using the TaqMan® chemistry genotyping system (Applied Biosystems, Foster City, CA, USA). All SNP genotyping assays were available and ordered using the Assay-on-Demand service from Applied Biosystems. These genotyping assays included primers to amplify the region containing the SNP of interest and two TaqMan Minor Groove Binder probes that are specific to the polymorphic variant alleles at the site labeled with different fluorescent 39 reporter dyes, FAM and VIC. All reactions were performed using standard conditions supplied by Applied Biosystems. Following thermocycling, fluorescence levels of the FAM and VIC dyes were measured and genotypes were scored using the Sequence Detection Systems 2.2 software (Applied Biosystems). Genotypes were uploaded into a Progeny database (Progeny Software, LLC, South Bend, IN, USA) containing the phenotypic data for subsequent statistical analysis. Using the following equation (Nyholt 2006), we determined that there was a >99% power to accurately differentiate between monozygotic (MZ) and dizygotic (DZ) twin pairs using >10 markers with minor allele frequencies between 0.2 and 0.5. 𝑀(𝐷𝑍) = 1 1 1 𝑃𝑟𝑜𝑏(𝑧0 ) + 𝑃𝑟𝑜𝑏(𝑧1 ) + 𝑃𝑟𝑜𝑏(𝑧2 ) 2 4 4 1 1 1 = 4 𝑀0 + 2 𝑎2 + 4 1 1 1 = 4 ∑𝑛𝑖=1(𝑝𝑖 )4 + ∑𝑛𝑖=1 ∑𝑛𝑗=𝑖+1(2𝑝𝑖 𝑝𝑗 )2 + 2 ∑𝑛𝑖=1(𝑝𝑖 )2 + 4 Of the 243 pairs, we identified 109 as DZ (r2 = 0.4-0.8) and 107 as MZ (r2>90%). All discordant sex twin pairs (N=165) were considered DZ. We excluded 27 pairs because of one or both twins having low genotyping efficiency that is <10 markers (N=18) or questionable zygosity that is r2<0.4 (N=5) or r2=0.81-0.9 (N=4). The utility of DNA extracted from DBS cards is challenging because of the very low DNA yield resulting from these small punches as starting material. This lower quality and quantity of DNA is most probably contributing to the low genotyping efficiency that we observed for some of the samples. Statistical Analysis Demographic characteristics were compared between DZ and MZ twin pairs. Chi square tests were used to compare categorical variables (gender, total parenteral nutrition, and abnormal screen) and Wilcoxon Rank sum tests were used to compare continuous traits (gestational age, weight, and age at screening). Heritability was estimated with 40 multilevel mixed-effects linear regression adjusting for confounders known to influence analyte measurements including gender, age at time of sample collection, gestational age and birth weight. P-values from the additive genetic component are presented. Heritability was estimated from the division of the beta coefficient of the additive component over the sum of the additive, shared environment and residual coefficients. Measurements outside the lower limits of detection (LOD) were given a value of LOD. 17-OHP, TSH and GALT had 10.9%, 5.6% and 0.1% of values, respectively, that were outside the LOD. GALT also had 59 (7.7%) measurements that were outside the upper limits of detection; these values were assigned as the upper limits. One amino acid (ASA) and eight acylcarnitines (C5:1, C6-DC, C14:2, C14-OH, C16:1-OH, C16-OH, C18-OH and C18:1-OH) had little to no variability (standard deviation ≤ 0.01) and, therefore, were excluded from heritability analysis. Succinylacetone was also excluded as >20% of the measurements were missing. For each analyte, outliers were excluded if values were outside of the mean±4 standard deviations. Equal numbers of DZ and MZ twin pairs were excluded for deviations ±4 standard deviations from the mean (P=0.58). The outliers may be due to neonatal illness that we could not account for since we did not have access to medical records. With the exception of C3, which had no outliers, 1-7 twin pairs (0.3%1.8%) were excluded for all other analytes. To normalize measurement distributions each analyte was transformed with the natural logarithm. Of the 37 analytes examined, 21 were normally distributed (P>0.01) after transformation; however MET and C18:1 required additional removal of 15 and 5 twin pair outliers respectively and results from these analytes should be interpreted accordingly. For ARG, LEU, PHE, VAL, C5, C10:1, C12:1, C14:1, C16:1, TSH and 17-OHP normal distribution was attained for the residuals from linear regression adjusting for relevant covariates including age at time of collection, birth weight, gestational age and gender. The residuals were used in heritability analyses for these analytes. TSH, PHE and C10:1 required additional removal of 36, 13 and 1 twin pair outliers respectively and results from these analytes should be 41 interpreted accordingly. GALT, C12 and C5-DC were analyzed with the natural log transformed measurement and IRT and C10 were analyzed with the residuals; however, all deviated from normality (p<0.01). Additionally we ran heritability measurements on natural log transformed analyte measurements excluding twin pairs <34 weeks gestation, with one or both twins having received an abnormal screen, with one or both twins on total parenteral nutrition and MZ twin pairs where the birth weight was >20% discordant. This was an attempt to control for infants with medical complications that could interfere with analyte measurements. As this was a population-based de-identified retrospective examination of data we were not able to connect our measurements to medical record information to obtain more detailed information on the severity or type of illness. Results were similar (Tables 2-3 and 2-4); therefore, the full model including all available twin pairs are presented in the results and discussion below. Analysis of Ratios There were eight amino acid ratios and 13 acylcarnitine ratios included in the measurements reported with the newborn screen (Table 2-1). We estimated heritability for these ratios as described above for the single analytes. C4/C2, C5/C2, C5-DC/C16 and C16-OH/C16 were excluded due to low variability (standard deviation ≤ 0.01). Outliers were excluded if values were outside of the mean±4 standard deviations; 1-7 twin pairs (0.3%-1.8%) were excluded for all ratios. Equal numbers of DZ and MZ twin pairs were excluded for deviations ±4 standard deviations from the mean (P=0.54). To normalize measurement distributions each ratio was transformed with the natural logarithm. Of the 17 analytes examined, 8 were normally distributed (P>0.01) after transformation; however ASP/HOMOCIT and LEU/PHE required additional removal of 5 and 2 twin pair outliers and results from these analytes should be interpreted accordingly. For C14:1/C12:1, CIT/ARG, TYR/PHE normal distribution was attained for the residuals from linear regression adjusting for relevant covariates including age at time 42 of collection, birth weight, gestational age and gender. The residuals were used in heritability analyses for these analytes. CIT/ARG required additional removal of 4 twin pair outliers respectively and results from these analytes should be interpreted accordingly. C4/C3 was analyzed with the natural log transformed measurement, C8/C10 was analyzed with the residuals, C5-DC/C8 was analyzed with the natural log transformed measurement after removal of 5 twin pairs and MET/PHE was analyzed with the residuals after removal of 2 twin pairs; however, all still deviated from normality (p<0.01) and should be interpreted with caution. Results Heritability analyses were performed on a total of 381 twin pairs (274 DZ and 107 MZ) (Table 2-5). The DZ and MZ twin pairs were not statistically different from each other when comparing their mean gestational age, total parenteral nutrition, abnormal newborn screening result, weight, and age at screening (Table 2-5). The gender of the two groups differed marginally between DZ and MZ twin pairs (P=0.06), with slightly more female MZ twin pairs compared to DZ (Table 2-5). Heritability measurements were significant after correction for multiple testing (p<0.001) for 10 analytes (Figure 2-1A, Table 2-3). C4-DC of the short chain acylcarnitines had the highest heritability (h2=0.83, P<10-16). Other short chain acylcarnitines with high heritability included C2 (h2=0.50, P=7x10-9), C3 (h2=0.44, P=2x10-6), C4 (h2=0.66, P=2x10-16),C4-OH (h2=0.31, P=4x10-5), and C5 (h2=0.61, P=1x10-9). Free carnitine C0 also had a significant heritability of (h2=0.45, P=4x10-9). The environmental component was significant (p<0.001) for all analytes except C4-DC and C5. However, in most cases where shared environment was also significant, the proportion of variability explained by the genetic component exceeded the proportion explained by shared environment. TSH (h2=0.58, P=2x10-5) and IRT (h2=0.52, P=3x10-9) also had a significant heritability measurement after correction for multiple testing. The 43 shared environment was significant for IRT (P=5x10-4) but not TSH (P=0.29). The heritability of 17-OHP (h2=0.31, P=1x10-3) was marginally significant with strong shared environment (P=7x10-7). The only amino acid significant after correction for multiple testing was glutamate (h2=0.35, P=5x10-4), the shared environment was also significant (P=1.0x10-5). There was a strong genetic component for the ratios PHE/TYR (h2=0.51, P=6.4x10-6), TYR/PHE (h2=0.59, P=6.6x10-7), C3/C2 (h2=0.64, P=8.7x10-13), C4/C3 (h2=0.61, P=2.9x10-12) and C5/C3 (h2=0.40, P=4.2x10-6) (Figure 2-1B, Table 2-6). Of special interest, are the results of the environment and genetic contribution to metabolites in the β oxidation pathway (Figure 2-2A). We observe a stronger environmental component for the variation in the long-chain and medium-chain acylcarnitine measurements. Whereas for short-chain acylcarnitines the environmental contribution decreases as the genetic contribution increases. We also observed the same trend of decreasing environmental and increasing genetic contribution for the pathway that represents the catabolism of the branched-chain amino acids (valine and total leucine) into C3-DC and C4-DC (Figure 2-2B). Discussion High-throughput technologies that integrate genomics and metabolomics are advantageous for providing insight into the pathophysiology and genetics of complex human diseases (Goonewardena et al. 2010). Hence, it is important to recognize the heritability of a particular metabolic measurement before investing in intensive genetic characterization. To our knowledge, this is the first evaluation of the heritability of a large panel of enzyme, acylcarnitine, and amino acid biomarkers in the neonatal period, in addition to being first to use newborn blood spot cards to estimate the genetic heritability. Our findings of high heritability for the acylcarnitines C2, C3, C4-OH and C5 as well as the amino acid glutamate were consistent with a previous report examining the heritability of metabolites in adults with a family history of premature cardiovascular 44 disease (Shah et al. 2009). These similarities are noteworthy not only because so few studies report heritability estimates for acylcarnitines, but also because there is recent and compelling evidence for the importance of acylcarnitines as biomarkers for cardiovascular disease, obesity, and type 2 diabetes in adulthood (Mihalik et al. 2010; Shah et al. 2010). Interestingly, our highest heritability was found with C4-DC which in addition to being elevated in diabetics, was also associated with poor glycemic control, implicating this metabolite as a strong biomarker for gluco- and lipotoxicity in type 2 diabetes (Mihalik et al. 2010). Furthermore, the same three metabolite classes (that is short-chain acylcarnitines, C4-DC, C4-OH) were reported to be significantly associated with adverse outcomes after coronary artery bypass grafting (Shah et al. 2012). Hence, these metabolites can be used as biomarkers of risk to predict performance after coronary artery bypass grafting (Shah et al. 2012). Our study suggests that the levels of potentially important biomarkers of adult diseases are heritable at birth. Therefore, identifying genetic variants associated with these metabolites at birth may provide important screening tools to identify diseases developed later in life. Also noteworthy is the significant finding of TSH heritability in neonates. TSH is one of the two analytes used in newborn screening to detect primary congenital hypothyroidism (CH) (Wilcken and Wiley 2008). As a major player in thyroid function, TSH has great influence on both neonatal as well as adult normal physiology, affecting almost all tissues and maintaining healthy status of all human systems including cognition, cardiovascular, skeletal, and metabolic function (Panicker 2011). Our finding of significant heritability for TSH in newborn twins (h2=0.58, P=2x10-5) agrees with previous reports of TSH heritability estimates in adult serum measurements, ranging from 0.32 (Samollow et al. 2004) to 0.64 (Hansen et al. 2004) to 0.65 (Panicker et al. 2008) in different studies. This finding of high TSH heritability, both in neonates as well as adults, 45 suggests that thyroid function is mainly controlled by genes throughout life, and hence, neonatal screening results of TSH might help predict adult thyroid diseases later in life. We also find relevance in these results with respect to neonatal metabolic screening programs. The State of Iowa Neonatal Metabolic Screening Program is one of the few that uses the C3/C2 ratio, for which we found strong heritability (h2=0.64, P=8.7x10-13), to screen for methylmalonic and propionic acidemias. We have identified patients with confirmed metabolic disorders based on elevated C3/C2 ratios in the presence of C3 below the threshold defined by the newborn screening program. This is consistent with the observation that the C3/C2 ratio has a strong genetic component with little environmental input. This suggests that the C3/C2 ratio may be a useful primary screening marker for methylmalonic and propionic acidemias as it is primarily defined by genetic influence. We are aware of the limitations in our study. Some of the studied metabolites had large confidence intervals; that may be due to our modest sample size. Furthermore, we were not able to connect our measurements to medical record information to obtain more detailed information on neonatal illness, we attempted to control for this by estimating the heritability excluding twin pairs <34 weeks gestation and twin pairs where one or both twins were on total parenteral nutrition and observed little difference compared to the model where all twin pairs were included (Table 2-3 and Table 2-4). Another limitation to our study was that we had no information on self-identified ancestry. However, in 2009 86.9% of the births in Iowa were Caucasian. However, it would be interesting for future studies to replicate these results in other races and age groups. We identified high heritability for TSH, and short-chain acylcarnitines all of which are important biomarkers of adult diseases. Our study illustrates the utility of having IRB reviewed access to stored, de-identified newborn dried bloodspot samples as these results will guide further studies addressing the predictive ability of metabolic biomarkers in 46 common complex adult diseases as well as characterizing the genes underlying these observed heritabilities. 47 Table 2-1 List of analytes examined. 17-Hydroxyprogesterone (17OHP-ng/mL) Acylcarnitines (cont) Galactose-1-Phosphate Uridyl Transferase (GALT-U/gHb) Tetradecenoylcarnitine (C14:1) Trypsinogen I Free (IRT-umol/L) Tetradecadienoylcarnitine (C14:2) Thyroid Stimulating Hormone (TSH-mIU/L) 3-Hydroxytetradecanoylcarnitine (C14-OH) Acylcarnitines (umol/L) Palmitoylcarnitine (C16) Carnitine free (C0) Palmitoleylcarnitine (C16:1) Acetylcarnitine (C2) 3-Hydroxypalmitoleylcarnitine (C16:1-OH) Propionylcarnitine (C3) 3-Hydroxypalmitoylcarnitine (C16-OH) Malonylcarnitine (C3-DC) Stearoylcarnitine (C18) Butyrylcarnitine+Isobutyrylcarnitine (C4) Oleoylcarnitine (C18:1) Methylmalonylcarnitine (C4-DC) 3-Hydroxyoleoylcarnitine (C18:1-OH) 3-Hydroxybutyrylcarnitine (C4-OH) Linoleoylcarnitine (C18:2) Isovalerylcarnitine+Methylbutyrylcarnitine (C5) 3-Hydroxystearoylcarnitine (C18-OH) Tiglylcarnitine (C5:1) Amino Acids (umol/L) Glutarylcarnitine (C5-DC) Alanine (ALA) 3-Hydroxyisovalerylcarnitine (C5-OH) Arginine (ARG) Hexanoylcarnitine (C6) Argininosuccinate (ASA) Methylglutarylcarnitine (C6-DC) Citrulline (CIT) Octanoylcarnitine (C8) Glutamate (GLU) Octenoylcarnitine (C8:1) Leucine (LEU) Decanoylcarnitine (C10) Methionine (MET) Decenoylcarnitine (C10:1) Phenylalanine (PHE) Dodecanoylcarnitine (C12) Succinylacetone (SUAC) Dodecenoylcarnitine (C12:1) Tyrosine (TYR) Tetradecanoylcarnitine (C14) Valine (VAL) 48 Table 2-2 List of markers used for zygosity testing and population allele frequencies. Chromosome Marker Position CEU CHB JPT YRI 1 rs581886 57539683 0.424 C 0.458 A 0.419 C 0.314 A 3 rs427832 149949053 0.491 G 0.226 G 0.273 G 0.372 A 4 rs3804099 154844106 0.45 C 0.339 C 0.267 C 0.372 T 4 rs8752 175649052 0.424 C 0.315 T 0.366 T 0.263 T 5 rs33383 142690179 0.481 T 0.247 C 0.2 C 0.41 C 5 rs4869315 96255028 0.455 A 0.494 A 0.372 A 0.451 G 6 rs2734335 32001923 0.487 G 0.387 G 0.151 G 0.496 G 8 rs2446432 67280862 0.492 A 0.378 C 0.375 C 0.333 C 8 rs4360312 4785152 0.46 T 0.217 C 0.198 C 0.265 T 10 rs2256588 81674775 0.483 A 0.5 G 0.467 A 0.2 G 10 rs4751996 118387884 0.431 A 0.284 G 0.256 G 0.44 A 11 rs1055574 17472409 0.482 G 0.274 G 0.291 G 0.389 G 12 rs2139573 101302904 0.474 C 0.267 C 0.295 C 0.3 C 17 rs2239907 23749871 0.46 T 0.271 T 0.221 T 0.451 C 17 rs4968779 58886303 0.5 G 0.125 G 0.25 G 0.217 A 19 rs1529729 11024562 0.473 C 0.256 C 0.174 C 0.279 C 21 rs6518251 45841556 0.487 T 0.476 T 0.448 T 0.363 T 22 rs738992 31540005 0.496 T 0.44 C 0.446 C 0.317 T 22 rs84460 35855677 0.335 G 0.363 G 0.372 G 0.442 G X rs2070584 47331463 0.473 T 0.458 G 0.43 G 0.424 G Table 2-3 Heritability estimates for all twin pairs. Enzyme h2 (95% CI) Genetic Coefficient Genetic Pvalue Environment Coefficient Environment Pvalue Standard h2 GALT 0.07 (2.0x10-3-0.77) 3.5x10-3 0.57 0.02 7.4x10-7 0.30 IRT 0.52 (0.35-0.68) 0.11 3.0x10-9 0.07 5.0x10-4 0.47 -3 0.20 -7 7.0x10 0.19 0.03 0.29 0.37 17-OHP 0.31 (0.16-0.51) 0.13 1.2x10 TSH 0.58 (0.32-0.80) 0.13 1.7x10-5 -3 -8 0.11 ALA 0.06 (1.1x10 -0.81) 0.01 0.61 0.05 2.4x10 ARG 0.22 (0.07-0.49) 0.07 0.05 0.15 1.9x10-7 0.10 -6 0.40 0.29 CIT 0.23 (0.08-0.51) 0.02 0.04 0.03 8.2x10 GLU 0.35 (0.18-0.56) 0.02 5.2x10-4 0.02 1.0x10-5 LEU -20 4.9x10 -16 MET 0.21 (0.05-0.54) PHE 3.2x10 -20 TYR 0.36 (0.16-0.63) -22 -18 (8.6x10 -2.8 x10 ) -22 -18 (9.4x10 -1.1x10 ) -8 -21 3.7x10 1.00 0.05 <1x10 0.02 0.10 0.05 2.6x10-5 -21 -15 1.3x10 1.00 0.02 8.7x10 0.06 5.5x10-3 0.05 4.5x10-3 -3 -14 -0.07 0.08 0.12 0.38 2.4x10-4 VAL 0.02 (7.8x10 -1.0) 1.3x10 0.87 0.06 2.4x10 C0 0.45 (0.31-0.60) 0.05 3.8x10-9 0.04 1.0x10-6 0.64 -9 0.04 -4 1.8x10 0.68 0.05 4.3x10-5 0.38 C2 0.50 (0.33-0.67) 0.05 6.6x10 C3 0.44 (0.27-0.61) 0.06 1.7x10-6 -10 0.13 (0.02-0.47) 0.02 0.21 0.09 2.6x10 C4 0.66 (0.49-0.80) 0.10 2.2x10-16 0.03 8.7x10-3 2.3x10-3 0.66 49 C3DC Table 2-3 Continued C4DC C4OH C5 C5DC C5OH C6 C8 C8:1 C10 C10:1 C12 C12:1 C14 C14:1 C16 0.83 (0.58-0.95) 0.31 (0.18-0.48) 0.61 (0.41-0.78) 0.03 (1.4x10 -1.0) -21 6.0x10 -13 -12 -8 -22 -19 (1.3x10 -1.4x10 ) (7.9x10 -1.3x10 ) (1.4x10 -2.5x10 ) 0.04 (4.4 x10 -0.83) 2.6x10 -14 -12 (1.3x10 -5.3x10 ) 0.31 (0.14-0.56) 0.14 (0.03-0.49) 0.27 (0.11-0.52) 1.1x10 1.4x10 0.02 0.03 0.24 4.0x10 -3 0.02 -16 -4 -13 0.10 -9 3.6x10 0.15 (0.04-0.44) 3.2x10 3.6x10 0.02 -6 -10 0.06 -5 0.02 0.20 (0.10-0.37) 4.2x10 <1x10-16 0.07 0.09 (0.02-0.39) -15 0.08 6.5x10 8.4x10 -11 8.7x10 -22 8.8x10 -3 9.9x10 -14 0.03 0.03 0.03 0.17 -3 0.84 0.09 1.00 0.11 1.00 0.17 1.00 0.09 0.66 0.13 1.00 6.6x10 0.06 0.07 0.13 -16 7.9x10-3 0.26 -3 0.20 0.04 0.11 0.01 0.04 0.39 0.70 -9 0.32 0.42 -16 0.06 5.6x10 -15 0.27 5.4x10 -8 -0.15 3.1x10 -11 -0.03 <1x10 -16 -0.10 <1x10 -16 -0.11 <1x10 -16 -0.28 -13 -0.10 -16 -0.08 1.0x10 -4 0.16 1.3x10 -8 -0.01 1.2x10 -5 0.24 -10 -1.0x10-3 <1x10 5.0x10 <1x10 C16:1 0.10 (0.01-0.51) 0.02 0.31 0.09 2.0x10 C18 0.31 (0.14-0.55) 0.03 5.1x10-3 0.04 5.2x10-5 0.39 0.03 -3 0.02 0.04 0.33 C18:1 C18:2 0.43 (0.21-0.68) 0.10 (0.01-0.51) 0.02 1.0x10 0.32 0.13 2.7x10 -11 0.19 50 The genetic coefficient and P-value represent the additive genetic component and the environment coefficient and P-value represent the shared environment component. Standard heritability was also calculated with the log transformed measurement and no covariate adjustment using 2*(rMZ-rDZ) where r is the standard correlation coefficient for MZ and DZ twins. 51 Table 2-4 Heritability estimates for twin pairs excluding TPN, abnormal screens, gestational age <34 weeks, and MZ twin pairs that are >20% discrepant in weight. Enzyme h2 (95% CI) Genetic Coefficient Genetic P-value Environment Coefficient Environment P-value GALT 2.8x10-13 (4.6x10-13-1.7x10-11) 1.52x10-14 1.00 0.03 <1x10-16 IRT 0.63 (0.41-0.8) 0.14 2.2x10-9 0.05 0.03 -6 0.11 2.7x10-5 OHP 0.43 (0.27-0.61) 0.12 1.6x10 TSH 0.5 (0.25-0.75) 0.11 4.4x10-4 0.05 0.09 0.36 0.04 3.8x10-6 -3 ALA 0.13 (0.01-0.63) 9.7x10 ARG 0.36 (0.18-0.59) 0.09 1.1x10-3 0.10 6.3x10-5 CIT 0.41 (0.19-0.67) 0.03 1.7x10-3 0.02 7.3x10-3 GLU 0.35 (0.16-0.6) 0.02 3.1x10-3 0.02 1.9x10-4 LEU 0.13 (0.01-0.68) 6.9x10-3 0.39 0.02 7.0x10-5 MET 0.28 (0.09-0.62) 0.02 0.05 0.03 1.9x10-3 PHE 0.14 (6.6x10-3-0.81) 5.3x10-3 0.48 0.01 0.02 TYR 0.47 (0.24-0.72) 0.07 5.3x10-4 0.04 0.04 VAL 0.26 (0.09-0.56) 0.02 0.04 0.03 4.3x10-5 C0 0.51 (0.35-0.66) 0.06 5.7x10-11 0.04 2.8x10-5 C2 0.52 (0.34-0.7) 0.06 2.4x10-8 0.04 1.1x10-3 C3 0.57 (0.37-0.75) 0.07 1.9x10-8 0.04 7.4x10-3 C3DC 0.18 (0.06-0.42) 0.03 0.04 0.10 1.5x10-10 C4 0.65 (0.45-0.81) 0.11 3.5x10-12 -16 0.04 0.01 -3 C4DC 0.87 (0.5-0.98) 0.09 6.7x10 C4OH 0.4 (0.25-0.56) 0.08 7.0x10-7 0.09 8.8x10-7 C5 0.69 (0.43-0.87) 0.07 1.4x10-8 0.01 0.29 C5DC 0.23 (0.12-0.38) 0.06 5.7x10-4 0.17 5.3x10-13 C5OH 0.28 (0.14-0.46) 0.03 8.6x10-4 0.06 4.3x10-9 C6 0.22 (0.05-0.63) 0.03 0.16 0.05 3.8x10-4 C8 0.34 (0.19-0.53) 0.06 9.2x10-5 0.09 1.3x10-7 C8:1 0.24 (0.14-0.38) 0.03 8.0x10-5 0.09 1.7x10-13 C10 0.34 (0.2-0.52) 0.09 6.9x10-5 0.14 1.2x10-7 C10:1 0.25 (0.12-0.45) 0.03 4.0x10-3 0.08 6.3x10-10 C12 0.22 (0.08-0.5) 0.04 0.04 0.10 3.4x10-7 5.1x10 0.65 52 Table 2-4 Continued C12:1 0.26 (0.13-0.44) 0.12 1.1x10-3 0.26 5.6x10-10 C14 0.42 (0.2-0.67) 0.05 1.4x10-3 0.03 7.2x10-3 C14:1 0.35 (0.19-0.56) 0.08 4.5x10-4 0.10 6.5x10-6 C16 0.37 (0.18-0.62) 0.04 2.2x10-3 0.04 9.5x10-4 C16:1 0.23 (0.09-0.48) 0.04 0.02 0.08 1.1x10-7 C18 0.42 (0.21-0.67) 0.05 6.6x10-4 0.03 4.8x10-3 C18:1 0.39 (0.17-0.68) 0.03 5.8x10-3 0.03 0.02 C18:2 0.28 (0.12-0.54) 0.05 0.01 0.09 4.0x10-6 The genetic coefficient and P-value represent the additive genetic component and the environment coefficient and P-value represent the shared environment component. 53 Table 2-5 Demographic characteristics of twin pairs. DZ (n=274) MZ (n=107) Gender of Twin Pair Pvalue 0.06* Female-Female 45 (16.4%) 58 (54.2%) Male-Female 165 (60.2%) 0 (0%) Male-Male 64 (23.4%) 49 (45.8%) Gestational Age of Twin Pair/weeks 35.6 (2.5) 35.7 (2.3) 1.00 TPN (one or both twins) 29 (10.6%) 9 (8.4%) 0.53 Abnormal screen (one or both twins) 42 (15.3%) 19 (17.8%) 0.56 Weight/grams (averaged for each twin pair) 2 397.9 (496.9) 2 364.7 (465.8) 0.19 Age at screening/hours (averaged for each twin pair) 35.2 (11.8) 35.2 (13.0) 0.55 TPN, total parenteral nutrition. *P-value determined comparing only Male-Male and Female-Female twin pairs. Table 2-6 Heritability estimates for analyte ratios. Enzyme h2 (95% CI) Genetic Coefficient Genetic value ARG/ORN 0.34(0.2-0.52) 0.06 ASP/HOMOCIT 0.33(0.13-0.62) CIT/ARG P- Environment Coefficient Environment P-value 6.1x10-5 0.08 3.4x10-8 0.03 0.01 0.03 3.8x10-3 0.17(0.04-0.47) 0.04 0.12 0.13 1.3x10-8 LEU/ALA 2.7x10-13(6.5x10-15-1.1x10-11) 1.4x10-14 1.00 0.03 0.00 LEU/PHE 0.17(0.04-0.46) 5.5x10-3 0.11 0.02 2.8x10-9 MET/PHE 0.04(1.8x10-5-0.99) 2.2x10-3 0.79 0.03 2.9x10-7 PHE/TYR 0.51(0.3-0.72) 0.09 6.4x10-6 0.04 0.03 -7 0.02 0.16 TYR/PHE 0.59(0.36-0.79) 0.10 6.6x10 C0/C16 0.14(0.03-0.43) 0.01 0.15 0.05 2.0x10-11 C0/C18 0.21(0.06-0.5) 0.02 0.07 0.04 9.5x10-7 C3/C2 0.64(0.45-0.79) 0.07 8.7x10-13 0.02 0.02 -12 0.04 6.5x10-3 C4/C3 0.61(0.43-0.76) 0.09 2.9x10 C5/C3 0.4(0.24-0.57) 0.05 4.2x10-6 0.05 4.1x10-6 C5DC/C8 0.11(0.03-0.35) 0.01 0.14 0.08 0.00 C8/C10 0.14(0.02-0.58) 6.8x10-3 0.29 0.02 1.3x10-5 C14:1/C12:1 0.06(1.0x10-3-0.77) 5.5x10-3 0.61 0.06 2.9x10-11 C14:1/C16 0.38(0.2-0.59) 0.05 3.8x10-4 0.05 1.1x10-4 54 The genetic coefficient and P-value represent the additive genetic component and the environment coefficient and P-value represent the shared environment component. 55 Figure 2-1 Heritability of analyzed metabolites and analyte ratios. (a) Heritability of analyzed metabolites. The y-axis is the negative log10 of the P-value for additive genetic component and the heritability estimate is on the x-axis. Confidence intervals around the heritability point estimates are in light gray bars. IRT, TSH, 17-OHP and GALT are represented as dark gray squares, amino acids are represented as medium gray circles and acylcarnitines are represented as light gray triangles. (b) Heritability of analyte ratios. The y-axis is the negative log10 of the P-value for additive genetic component and the heritability estimate is on the x-axis. Confidence intervals around the heritability point estimates are in light gray bars. 56 Figure 2-2 Environment and genetic contribution to metabolites in the β-oxidation pathway and the pathway for catabolism of branched-chain amino acids. (a) Environment and genetic contribution to metabolites in the β-oxidation pathway. The x-axis is a list of the even-chain acylcarnitines, the y-axis is the negative log10 of the P-value for the correlation coefficients. The dotted line represents the genetic component and the solid line represents the environmental component. (b) Environment and genetic contribution to metabolites along the pathway that represents the catabolism of the branched-chain amino acids (valine and total leucine) into C3-DC and C4-DC. The x-axis is a list of the metabolites, the y-axis is the negative log10 of the P-value for the correlation coefficients. The dotted line represents the genetic component and the solid line represents the environmental component. 57 CHAPTER 3 GENETIC ASSOCIATIONS WITH NEONATAL THYROID STIMULATING HORMONE LEVELS The following manuscript was published online on February 27, 2013 in Pediatric Research. Additional authors include Oleg Shchelochkov, Stanton Berberich, Jeffrey Murray, and Kelli Ryckman. Thyroid stimulating hormone (TSH) is known to be largely controlled by genetic factors. In the previous chapter, we presented that TSH heritability in neonates was 58%. Genetic factors playing a role in regulating TSH levels have been extensively studied in adults through genome-wide association and candidate-gene studies. However, the genetic factors involved in neonatal TSH variability have never been studied. In this study, we identified for the first time two genetic associations with neonatal TSH levels that replicate findings with adult TSH levels. This finding may have clinical implications for the early prediction of risk for adult diseases and conditions associated with thyroid hormone levels. Dr. Oleg Shchelochkov provided valuable insight into TSH use as a marker for congenital hypothyroidism and was involved in editing the manuscript. Dr. Stanton Berberich provided insight on the study design and study samples and data. Drs. Jeffrey Murray and Kelli Ryckman provided oversight for all aspects of the project, provided input into study design and assisted in the writing and editing of the manuscript. Dr. Kelli Ryckman also performed the statistical analyses. I reviewed the literature and selected all study markers. I performed part of the DNA extraction and trained Dr. Osayame Ekhaguere, Lauren Fleener, and Daniel Cook to complete additional DNA extraction from newborn blood spot cards. I optimized a new protocol for Fluidigm genotyping that had not been used in our lab before and performed all the genotyping presented in this 58 study. I performed bioinformatic analyses and assisted in statistical analyses. I also wrote the manuscript, revisions, and responses to reviewer critiques. Abstract Background: Elevations or deficits in thyroid hormone levels are responsible for a wide range of neonatal and adult phenotypes. Several genome-wide, candidate-gene and meta-analysis studies have examined thyroid hormones in adults; however, to our knowledge no genetic association studies have been performed with neonatal thyroid levels. Methods: A population of Iowa neonates; term (n=827) and preterm (n=815), were genotyped for 45 single nucleotide polymorphisms. Thyroid stimulating hormone (TSH) values were obtained from the Iowa Neonatal Metabolic Screening Program. Analysis of variance was performed to identify genetic associations with TSH concentrations. Results: The strongest association was rs4704397 in the PDE8B gene (p=1.3x10-4), followed by rs965513 (p=6.4x10-4) on chromosome 9 upstream of the FOXE1 gene. Both of these SNPs met statistical significance after correction for multiple testing. Six other SNPs were marginally significant (p<0.05). Conclusions: We demonstrated for the first time two genetic associations with neonatal TSH levels that replicate findings with adult TSH levels. These SNPs should be considered as early predictors of risk for adult diseases and conditions associated with thyroid hormone levels. Furthermore, this provides a better understanding of the thyroid profile and potential risk for thyroid disorders in newborns. Introduction Endocrine disorders are substantial contributors to neonatal morbidity and mortality and, of these; congenital hypothyroidism (CH) is the most common (Kumar et al. 2009). CH is a common and preventable cause of mental retardation with an incidence rate of approximately 1 in 2,350 live births (Hinton et al. 2010). Early treatment with thyroxine (T4) with subsequent supplementation for life produces excellent results for 59 both growth and development (Wilcken and Wiley 2008; Hinton et al. 2010). CH is screened for at birth through detection of T4, thyroid stimulating hormone (TSH), or both (Wilcken and Wiley 2008). In the preterm infant, thyroid function undergoes postnatal changes related to an immature hypothalamic-pituitary axis (HPA), along with the interrupted exposure to maternal thyroid hormone and thyroid releasing hormone from the placenta. Due to immature HPA function T4 is lower in preterm infants compared to neonates born at term, and there is a direct correlation between the serum T4 level and the degree of prematurity (LaFranchi 1999). Preterm neonates with abnormal thyroid function may have transient hypothyroxinemia of prematurity, and be misreported as true cases of CH (Hinton et al. 2010). Hence, it is essential to take gestational age and birth weight into consideration when making the differentiation between transient hypothyroidism and true cases of CH (Hinton et al. 2010). Among healthy subjects, TSH shows considerable variability between individuals, whereas this variability is much less in the same individual when TSH is measured repeatedly over an extended period of time (Panicker et al. 2010). Previous studies have observed that TSH variability is under strong genetic regulation; studies have estimated heritability of up to 65% for variation in adult serum TSH (Panicker et al. 2008). In addition, there have been several genome-wide association studies (GWAS) reporting multiple genetic variants associated with TSH levels in adults (Hwang et al. 2007; Arnaud-Lopez et al. 2008; Panicker et al. 2010; Rawal et al. 2012). Furthermore in candidate-gene studies the Asp727Glu polymorphism in the TSH receptor gene (TSHR) is associated with adult serum TSH levels, which further supports a genetic contribution in assessing the variation of TSH (Hansen et al. 2007). To our knowledge no studies have examined candidate SNPs with neonatal TSH measurements. We genotyped term and preterm infants for 45 SNPs in 24 candidate genes that are known to play a role in TSH production or metabolism and examined these polymorphisms for associations with TSH 60 levels measured at birth as part of the Iowa Neonatal Metabolic Screening Program. Understanding variation in TSH levels and the genes responsible may be particularly important in a population at risk for abnormal TSH levels such as preterm infants. Methods Study Population Study samples were obtained from two sources. The first was from the University of Iowa SHL where we obtained de-identified residual dried blood spots on a population of Iowa neonates; term (n=827) and preterm (n=413). Approval for use of the deidentified data and blood spot cards was granted by the Iowa Department of Public Health and a waiver of consent was obtained from the Institutional Review Board at the University of Iowa (IRB#200908793). All subjects had TSH measurements between day 1 and 3 of life and had not received a transfusion prior to sample collection. Preterm birth was defined as birth before 37 completed weeks of gestation. Gestational age was determined from the records obtained from the SHL. Quantification of TSH was performed at the SHL using CLIA (Clinical Laboratory Improvement Amendments) certified methods. TSH was determined by solid phase, time-resolved fluoroimmunoassay from dried newborn blood spots using PerkinElmer’s AutoDELFIA platform (Waltham, MA). DNA was extracted from one dried whole blood spot for all term samples and two dried whole blood spots for all preterm samples using the AutoGen (Holliston, MA) QuickGene-810 nucleic acid extraction machine with the DNA Tissue Kit (AutoGen) and following manufacturer’s recommendations. The second source was from the University of Iowa Hospitals and Clinics NICU. Preterm infants admitted to this NICU were either born in house or transferred from referring units within the first 28 days of life. Preterm birth was also defined as birth before 37 completed weeks of gestation. Gestational age was determined from the first day of the last menstrual period. Gestational age was confirmed through obstetrical 61 judgment and if uncertain, ultrasound measurements were used for confirmation. All families provided signed informed consent (UI IRB199911068) to be included in a repository of samples designed to provide DNA and limited epidemiologic data to study complications of newborn infants (Steffen et al. 2007). Preterm infants from singleton births, without congenital anomalies, with gestational ages between 22 to 36 weeks (n=402) were included in this study, and early preterm infants (<34 weeks gestation) were overrepresented from their population frequency due to the referral patterns of our NICU. TSH measured by the SHL as part of routine neonatal screening was linked to the sample and medical record data. DNA was extracted from cord blood, discarded venous blood or buccal swab for all infants. All DNA was extracted using standard protocols. Marker Selection and Genotyping We chose a total of 45 SNPs encompassing 24 candidate genes and eight intergenic loci. The list of genes and SNPs genotyped are in Table 3-1. SNPs were chosen either based on their previously reported associations with adult serum TSH levels, or based on thyroid biology and candidate genes from the literature. In the first case, we chose the exact SNPs that were reported to be associated. In the second case, we chose SNPs from the HapMap database (http://hapmap.ncbi.nlm.nih.gov/) based on their ability to tag the surrounding SNPs in the candidate gene. All SNPs were genotyped using TaqMan assays (Applied Biosystems, Foster City, CA) on the EP1 SNP Genotyping System and GT48.48 Dynamic Array Integrated Fluidic Circuits (IFCs) (Fluidigm, San Francisco, CA). All SNP genotyping assays were available and ordered using the Assay-on-Demand service from Applied Biosystems. These genotyping assays included primers to amplify the region containing the SNP of interest and two TaqMan Minor Groove Binder probes that are specific to the polymorphic variant alleles at the site labeled with different fluorescent reporter dyes, FAM and VIC. All reactions were performed using standard conditions supplied by Fluidigm. Following thermocycling, 62 fluorescence levels of the FAM and VIC dyes were measured using the EP1 Reader (Fluidigm) and genotypes were scored using the Fluidigm Genotyping Analysis software (Fluidigm). Genotypes were uploaded into a Progeny database (Progeny Software, LLC, South Bend, IN) containing the phenotypic data for subsequent statistical analysis. Genotyping efficiency was >98% for all markers with the exception of rs9322817 in HACE1 (93.5%) and rs1014968 in TSHR (90.8%). Forty-three individuals with genotyping efficiency <95% (i.e. missing data on >2 markers) were excluded from further analysis. Additionally, only 16 individuals had an abnormal (TSH≥25.0 mIU/L) level on initial screen. In order to avoid potential confounding, these individuals were excluded leaving 404 SHL preterm neonates, 799 term neonates and 380 NICU neonates for analysis. Statistical Analysis Demographic characteristics were compared between cohorts using Chi-square tests for categorical traits and Wilcoxon rank sum tests for continuous traits. All demographic factors listed in Table 3-2, year of sampling and TSH assay lot were individually associated (P≤0.001) with the natural log transformed TSH measurement in the combined (N=1,583) sample cohort using analysis of variance (ANOVA). Step-wise modeling was used to determine a final list of covariates for inclusion when testing association between SNP genotype and TSH level. Gestational age (P=5.55x10-19), age at time of sampling (P=1.63x10-42), gender (P=1.52x10-8) and year of sampling (P=7.36x104 ) were all significantly associated with TSH in the final model; birth weight (P=0.02), assay lot (P=0.09) and TPN (P=0.25) were not included in the final model due to marginal significance. Hardy-Weinberg Equilibrium (HWE) was evaluated for each marker using Fisher exact tests for all cohorts. All markers were tested for association with the natural log transformed TSH measurement adjusting for gender, gestational age, age at time of sampling and year of sampling using ANOVA in the combined cohort 63 (N=1,583). A Bonferonni significance threshold of P=0.001 (0.05/45 markers) was used to correct for multiple testing. Sub-analyses were performed in each cohort separately to determine individual population effects. All analyses were performed in STATA software version 12.0 (StataCorp LP, College Station, TX). Haplotype Analysis Haplotype analysis of SNPs in the same gene or region was used to evaluate regional associations with natural log transformed TSH levels. Haplotype analysis was performed with linear regression adjusted for gender, gestational age, age at time of sampling and year of sampling in the combined cohort. Haplotype analysis was performed using sliding windows of 2, 3, and 4 SNPs along a gene or region of interest. Haplotype analyses were performed in PLINK software (http://pngu.mgh.harvard.edu/purcell/plink/) (Purcell et al. 2007). Results Analysis was performed on a total of 1,583 neonates (404 University of Iowa State Hygienic Laboratory (SHL) preterm neonates, 799 term neonates, and 380 University of Iowa Hospitals and Clinics Neonatal Intensive Care Unit (NICU) neonates) (Table 3-2). The three cohorts were not statistically different from each other when comparing their gender (Table 3-2). Differences between mean TSH level, gestational age, and birth weight were statistically significant between all three cohorts (Table 3-2). Difference in mean for age at screening was statistically significant between SHL preterm and NICU preterm but not between SHL term and SHL preterm cohorts (Table 3-2). Difference in total parenteral nutrition was statistically significant between SHL term and SHL preterm but not between SHL preterm and NICU preterm cohorts (Table 3-2). Association results for all genotyped single nucleotide polymorphisms (SNPs) in combined populations are shown in Figure 3-1 and Table 3-3. Association results for all genotyped SNPs in individual populations are shown in Figure 3-2 and Table 3-4. 64 Eight SNPs were nominally significantly associated with neonatal TSH levels in all study populations combined (p<0.05) (Table 3-5, Table 3-6, and Figure 3-1).The strongest association was rs4704397 in the PDE8B gene (p=1.3x10-4), followed by rs965513 (p=6.4x10-4) on chromosome 9 upstream of the FOXE1 gene. Both of these SNPs met statistical significance after correction for multiple testing (corrected significance threshold set at p<0.001). While none of the associations in the individual populations met the significance threshold after correction for multiple testing, the strongest associations remained rs4704397 in the PDE8B and rs965513 near FOXE1 (Figure 3-2). Each copy of the minor A allele for rs4704397 in PDE8B was associated with an increase in TSH levels in both term and preterm infants (Table 3-5), as well as an increase of 0.6 mIU/L in TSH levels in the combined cohort. For SNP rs965513 in FOXE1 each copy of the minor A allele was associated with a decrease in TSH levels in both term and preterm infants (Table 3-5), as well as a decrease of 0.2-0.7 mIU/L in TSH levels in the combined cohort. Two other SNPs in FOXE1 (rs1443432 and rs3021523) showed marginal (p<0.05) significance with TSH levels. Each copy of the G allele of rs1443432 and each copy of the C allele of rs3021523 were associated with a decrease in TSH levels in both term and preterm infants (Table 3-5), as well as a decrease of 0.2-0.5 and 0.2-0.6 mIU/L in TSH levels in the combined cohort respectively. Haplotype analysis revealed significant associations of specific allele combinations with TSH levels. Near the FOXE1 gene, the presence of the AT and GT haplotypes at rs965513 and rs1443433 was significantly associated with either a decrease or increase of TSH levels, respectively (p=4.2x10-4 and p=4.6x10-4) (Table 3-7). The four remaining SNPs were marginally significant with TSH levels; rs9308765 on chromosome 2 (p=0.01), rs657152 in the ABO gene (p=0.02), rs4892386 in the SLC16A2 gene (p=0.01), and rs1527680 in the PPP1R9A gene (p=0.04). However, the association between these SNPs and TSH levels were not always in the same direction in the three studied cohorts, and further investigation is needed to confirm the associations 65 (Table 3-6). While there were no significant single locus associations with TSH levels in TSHR in any of the cohorts the presence of the AGT haplotype at rs10149689, rs4903957, and rs11159483, the GTT haplotype at rs4903957, rs11159483, and rs2075179, the AGTT haplotype at rs10149689, rs4903957, rs11159483, and rs2075179, and the GTTA haplotype at rs4903957, rs11159483, rs2075179, and rs12885526 were significantly associated with TSH levels (p=7.9x10-4, p=6.2x10-4, p=4.2x10-5, and p=7.8x10-4 respectively) (Table 3-8). Discussion Elucidating the genetic basis of TSH variability is currently an area of interest to further the understanding of adult conditions related to TSH levels. For example, low serum TSH levels are associated with an increased risk of atrial fibrillation in adults over 60 years old (Sawin et al. 1994). Thyroid hormone levels within the normal physiological range have also been shown to affect bone mass and density in healthy men aged 25-45 years (Roef et al. 2011), as well as in men and women above 55 years of age (van der Deure et al. 2008). Thyroid hormones also play a key role in neonatal as well as adult normal physiology, affecting almost all tissues and maintaining healthy status of all human systems including cognition, cardiovascular function, skeletal health, and balanced energy and metabolic status (Panicker 2011). This is particularly relevant in preterm infants were the variability of TSH is particularly high because of postnatal changes in thyroid function due to premature interrupted exposure to maternal thyroid hormones as well as an immature HPA (Hinton et al. 2010). Understanding the shared genetic associations with TSH levels in both the neonatal period as well as through adulthood will be useful for earlier prediction of risk to adult diseases that are affected by TSH levels. In this light, we aimed to identify genetic polymorphisms that may play a role in TSH variation, especially, in preterm infants. We observed genetic associations that have 66 an effect on TSH levels in either or both term and preterm infants. However, most of our nominally significant associations were found in the term population, and these associations reached multiple testing correction levels of significance in all populations combined. This may be due to the wide range of variability in TSH levels of preterm infants due to immature HPA and the skewed nature of our preterm population where early preterm infants are overrepresented. Our most significant association was with SNP rs4704397 in PDE8B gene (p=1.3x10-4). Each copy of the minor A allele was associated with an increase of 0.6 mIU/L in TSH levels in the combined cohort. This finding is consistent with what has previously been reported in adult serum TSH levels (ArnaudLopez et al. 2008). The PDE8B gene is expressed most abundantly in the thyroid gland where it has threefold higher levels than in the next highest tissue (Lakics et al. 2010). It is expressed at lower levels in some other tissues including the brain, spinal cord, and placenta (Hayashi et al. 1998). The PDE8B gene encodes a high affinity adenosine 3’,5’cyclic monophosphate (cAMP)-specific phosphodiesterase to regulate the level of cAMP in cells and plays a vital role in signal transduction (Hayashi et al. 1998; Lakics et al. 2010). Common genetic variants in PDE8B may affect steroid hormone physiology, such as levels of TSH. For example, an intronic SNP in PDE8B (rs4704397) that was identified in the genome-wide association study by Arnaud-Lopez et al. (2008) associated with adult serum TSH levels was then reported to be associated with subclinical hypothyroidism during pregnancy (Shields et al. 2009), as well as with TSH levels in obese children (Grandone et al. 2012). Interestingly, we found the same SNP to be associated with TSH levels in both preterm and term infants suggesting that this gene plays an important role in regulating TSH levels at birth as well as thyroid function throughout life. All four studies (adult serum TSH level (Arnaud-Lopez et al. 2008), pregnancy TSH level (Shields et al. 2009), obese children TSH level (Grandone et al. 2012), and this newborn TSH level study) came to the same finding that each copy of the minor A allele was associated with an increase in TSH levels. In the Arnaud-Lopez et al. 67 paper, the authors suggest that since cAMP is necessary for thyroid-hormone secretion due to TSH stimulation; when PDE8B catalyzes the hydrolysis and inactivation of cAMP in the thyroid gland, it results in decreased generation of thyroid-hormone T4 and T3 resulting in the negative feedback loop to act on producing more TSH (Arnaud-Lopez et al. 2008). Hence, genetic variation in PDE8B may affect PDE8B activity resulting in altered cAMP, TSH, and probably other downstream effects. In the Arnaud-Lopez et al. paper, all exons in the PDE8B gene were sequenced in 40 patients to identify a possible etiologic variant in linkage disequilibrium with the intronic rs4704397; however, no coding variants were identified (Arnaud-Lopez et al. 2008). Further investigation and sequencing of this gene will be needed in adult and newborn samples to identify the regulatory regions causing the association between PDE8B and thyroid levels. We also found SNPs rs965513 and rs1443432 near the FOXE1 gene (p=6.4x10-4 and 0.02 respectively) and SNP rs3021523 in the FOXE1 gene (p=0.03), to be associated with TSH levels in all the populations combined. Although, only rs965513 meets multiple testing correction level of significance while the other two SNPs are only marginally significant, this may be secondary to the moderate, but not complete, linkage disequilibrium (LD) with each other (rs965513, rs1443432: D’=0.863, r-squared =0.519; rs965513, rs3021523: D’=0.835, r-squared=0.48) and where rs965513 and rs1443433 also have strong effects as part of a haplotype associated with TSH levels. FOXE1 gene encodes a transcription factor that is essential for the initiation of thyroid differentiation at the embryonic stage (Parlato et al. 2004). Mutations of the FOXE1 gene may result in thyroid dysgenesis leading to both familial as well as cases of syndromic congenital hypothyroidism in the Bamforth-Lazarus Syndrome, Online Mendelian Inheritance in Man (OMIM) #241850 (Online Mendelian Inheritance in Man 2013), a rare inherited disorder characterized by CH, cleft palate, and spiky hair (Castanet and Polak 2010). FOXE1 also plays an important role in regulating the transcription of different thyroidspecific genes resulting in regulation of thyroid-hormone synthesis (Gudmundsson et al. 68 2009). Both of two SNPs, rs965513 near FOXE1 (Castanet and Polak 2010) and rs1443434 in FOXE1 gene (Medici et al. 2011), have been previously shown to be associated with adult serum TSH levels. Our replication of this finding with newborn TSH levels further implicates involvement of this locus in determining TSH levels in newborns; indicating that this association is present at birth. We further identified four haplotypes in the TSHR gene to be significantly associated with TSH levels. The protein encoded by the TSHR gene is the TSH receptor (TSHR). TSHR is present on thyroid cells, and when activated by TSH secreted from the pituitary gland, intracellular cAMP is upregulated resulting in activation of various cellular processes ending with an increased production of thyroid hormone (Stathatos 2012). SNPs in the TSHR gene were previously found to be associated with adult serum TSH levels in a GWAS reported by Arnaud-Lopez et al. (2008). We did not find an association with single SNPs in the TSHR gene; however we found SNP haplotypes to be associated with newborn TSH levels. One limitation of this study is that we were not able to connect TSH measurements to medical record information to obtain more detailed information on neonatal illness or race for the SHL preterm and term cohorts. However, in 2009 86.9% of the births in Iowa were Caucasian, suggesting that the majority of our samples are from Caucasians. Another limitation was that we do not have follow up data on our study cohort to check for development of conditions later in life. On the other hand, our study illustrates the utility of having IRB reviewed access to stored, de-identified newborn dried bloodspot samples for genetic studies. It is also, to our knowledge, the first study to examine genetics of TSH levels in newborns. Unraveling the genes that affect TSH levels will enhance our understanding of the genetic regulation and physiology of the thyroid and the pituitary-thyroid axis as well as the genes that may be involved in different thyroid diseases. This knowledge may be used to help individualize thyroid related treatments according to the individual’s specific genotype. Recognizing TSH variation 69 and the genetic factors affecting it early in infancy may be a useful adjunct in population screening for the early prediction and possible treatment or prevention of adult diseases and conditions affected by TSH levels. 70 Table 3-1 List of genotyped markers. Marker Gene Location Chr. Position MAF rs10917469 - Intergenic 1 19843576 0.19 rs1321108 TSHB Promoter 1 115572365 0.5 rs4849179 PAX8 Intron 2 113985170 0.42 rs877202 PAX8 Intron 2 114019129 0.26 rs9308765 - Intergenic 2 119043209 0.11 rs2288629 EPHA4 Intron 2 222307310 0.16 rs1505287 THRB Intron 3 24412690 0.37 rs784490 TTC21A Intron 3 39173530 0.24 rs1976324 - Intergenic 3 87212806 0.31 rs10493147 HSPA4L Intron 4 128737499 0.24 rs2545308 - Intergenic 4 181637915 0.44 rs27178 PDE4D Intron 5 58587025 0.46 rs4704397 PDE8B Intron 5 76518442 0.48 rs9342104 CGA Intron 6 87798512 0.44 rs6942231 HACE1 Intron 6 105191814 0.45 rs9322817 HACE1 Intron 6 105232233 0.4 rs2983521 PDE10A Intron 6 166057203 0.2 rs10486365 TMEM196 Intron 7 19801364 0.1 rs6977660 TMEM196 Intron 7 19805480 0.14 rs10499559 - Intergenic 7 22109459 0.16 rs10486653 NPSR1 Intron 7 34711663 0.2 rs1527680 PPP1R9A Promoter 7 94534886 0.15 rs2252696 SLA Intron 8 134063532 0.43 rs7865184 ZDHHC21 Intron 9 14687867 0.45 rs10512065 GNAQ Intron 9 80625644 0.13 rs965513 - Intergenic 9 100556109 0.37 rs1443433 - Intergenic 9 100579219 0.19 rs1443432 - Intergenic 9 100583195 0.47 rs894673 FOXE1 Promoter 9 100612270 0.47 rs3021523 FOXE1 Coding exon-synon 9 100616583 0.29 rs4460498 FOXE1 Downstream 9 100620412 0.46 rs4128956 MED27 Intron 9 134818509 0.38 71 Table 3-1 Continued rs657152 ABO Intron 9 136139265 0.38 rs944289 - Intergenic 14 36649246 0.41 rs10149689 TSHR Promoter 14 81415800 0.46 rs4903957 TSHR Intron 14 81448511 0.32 rs11159483 TSHR Intron 14 81501311 0.25 rs2075179 TSHR Coding exon-synon 14 81562998 0.1 rs12885526 TSHR Intron 14 81574429 0.36 rs1991517 TSHR Coding exon-missense 14 81610583 0.1 rs945006 DIO3 3'UTR 14 102029277 0.12 rs3908399 - Intergenic 20 12901275 0.27 rs6030171 PTPRT Intron 20 40994094 0.31 rs500243 SLC16A2 Intron X 73676685 0.37 rs4892386 SLC16A2 Intron X 73718735 0.32 Chr: chromosome, MAF: minor allele frequency, UTR: untranslated region. 72 Table 3-2 Demographic Characteristics of Cohorts. a SHL term (N=799) SHL preterm (N=404) NICU preterm (N=380) P-valuea P-valueb TSH level (mIU/L) 8.9±4.5 7.4±4.5 5.9±3.8 <0.001 <0.001 Male Gender 454 (56.8%) 230 (56.9%) 222 (58.4%) 0.97 0.67 Gestational Age (weeks) 40.0±0 34.0±2.6 31.2±3.1 <0.0001 <0.0001 Birth weight (grams) 3,489.9±434.0 2,405.1±695.3 1,773.4±712.3 <0.0001 <0.0001 Age at screening (hours) 36.4±10.6 37.6±12.8 29.5±7.0 0.68 <0.0001 Total parenteral nutrition 2 (0.3%) 51 (12.6%) 38 (10.0%) <0.001 0.25 P-values for differences between SHL term and SHL preterm. b P-values for differences between SHL preterm and NICU preterm. 73 Table 3-3 Association statistics for all genotyped SNPs with neonatal TSH level for all combined populations. P-values reported in this table are from ANOVA and nonparametric analysis. SHL Combined (N=1,203) Preterm Combined (N=784) All Populations (N=1,583) SNP ANOVA NP ANOVA NP ANOVA NP rs10917469 0.7528 0.3101 0.2791 0.2005 0.9458 0.7193 rs1321108 0.9158 0.6954 0.303 0.7759 0.4175 0.8148 rs4849179 0.2598 0.4742 0.2782 0.481 0.2721 0.1839 rs877202 0.5271 0.6602 0.165 0.4215 0.1831 0.4164 rs9308765 0.052 0.3822 0.0841 0.2538 0.0142 0.1066 rs2288629 0.1125 0.45 0.224 0.4296 0.048 0.8108 rs1505287 0.6627 0.3227 0.649 0.9471 0.4997 0.2211 rs784490 0.4193 0.2856 0.1671 0.2018 0.5526 0.528 rs1976324 0.3268 0.6366 0.8745 0.8343 0.8638 0.9436 rs10493147 0.546 0.358 0.0701 0.0443 0.1836 0.1079 rs2545308 0.9677 0.8922 0.5966 0.5533 0.7198 0.9349 rs27178 0.9619 0.8553 0.2537 0.1269 0.5674 0.3666 rs4704397 0.0012 0.00037 0.0032 0.0074 0.000131 0.000198 rs9342104 0.6594 0.8155 0.4234 0.4766 0.9504 0.8611 rs6942231 0.0812 0.539 0.369 0.1698 0.6181 0.4383 rs9322817 0.0609 0.438 0.2374 0.1315 0.3853 0.2407 rs2983521 0.2517 0.3254 0.6611 0.7188 0.4708 0.7915 rs10486365 0.3561 0.1761 0.8154 0.6479 0.8889 0.2256 rs6977660 0.2457 0.1685 0.0844 0.1737 0.266 0.0699 rs10499559 0.7201 0.6735 0.8018 0.5151 0.6427 0.4651 rs10486653 0.7473 0.5105 0.3012 0.5109 0.8398 0.9275 rs1527680 0.2204 0.1149 0.041 0.0707 0.0408 0.0154 rs2252696 0.863 0.8943 0.9006 0.4825 0.8176 0.6244 rs7865184 0.4494 0.6661 0.7593 0.1664 0.9355 0.4005 rs10512065 0.3765 0.08 0.062 0.0229 0.3937 0.047 rs965513 0.002 0.0237 0.0163 0.0786 0.00064 0.0039 rs1443433 0.8521 0.8393 0.666 0.5086 0.8468 0.61 rs1443432 0.0466 0.0874 0.2357 0.451 0.0229 0.0285 rs894673 0.2178 0.4816 0.8799 0.851 0.2518 0.6393 rs3021523 0.0884 0.0927 0.0437 0.1423 0.0324 0.047 74 Table 3-3 Continued rs4460498 0.1777 0.406 0.7991 0.8849 0.1892 0.5 rs4128956 0.0679 0.2111 0.366 0.6094 0.1854 0.7661 rs657152 0.0493 0.1728 0.0928 0.2228 0.0173 0.0742 rs944289 0.4915 0.3661 0.6303 0.7263 0.8397 0.8868 rs10149689 0.3957 0.2723 0.216 0.4152 0.1642 0.2357 rs4903957 0.6061 0.1911 0.8394 0.6728 0.4663 0.3086 rs11159483 0.2454 0.2215 0.1874 0.09 0.1936 0.1026 rs2075179 0.7826 0.9824 0.8474 0.9097 0.7666 0.9981 rs12885526 0.559 0.4342 0.9649 0.7441 0.7106 0.831 rs1991517 0.7395 0.9231 0.0433 0.1712 0.3744 0.6739 rs945006 0.8651 0.2766 0.267 0.5691 0.7954 0.61 rs3908399 0.044 0.1776 0.8479 0.3134 0.3641 0.2268 rs6030171 0.8891 0.6489 0.5915 0.6862 0.7144 0.4469 rs500243 0.128 0.0756 0.4275 0.797 0.1428 0.0968 rs4892386 0.0073 0.0095 0.0582 0.2294 0.0143 0.0583 75 Table 3-4 Association statistics for all genotyped SNPs with neonatal TSH level for individual population cohorts. P-values reported in this table are from ANOVA and nonparametric analysis. SHL Term (N=799) SHL Preterm (N=404) NICU Preterm (N=380) SNP ANOVA NP ANOVA NP ANOVA NP rs10917469 0.348 0.0724 0.4504 0.534 0.511 0.3517 rs1321108 0.9127 0.6842 0.9386 0.6803 0.0795 0.1883 rs4849179 0.5091 0.3613 0.4075 0.9425 0.4809 0.4565 rs877202 0.5594 0.5593 0.5057 0.971 0.2645 0.2225 rs9308765 0.0358 0.1824 0.2488 0.6247 0.2044 0.1287 rs2288629 0.2654 0.6044 0.643 0.2896 0.3443 0.6087 rs1505287 0.5449 0.2636 0.5706 0.4703 0.597 0.3648 rs784490 0.5856 0.2744 0.1172 0.1133 0.6059 0.6838 rs1976324 0.4701 0.6724 0.1757 0.1484 0.078 0.1744 rs10493147 0.9838 0.8336 0.306 0.173 0.2324 0.1562 rs2545308 0.6793 0.7623 0.8728 0.8949 0.5124 0.3412 rs27178 0.7575 0.762 0.9138 0.5949 0.1517 0.2181 rs4704397 0.0184 0.000578 0.0571 0.0677 0.0369 0.0658 rs9342104 0.443 0.7448 0.9974 0.956 0.1363 0.1017 rs6942231 0.1881 0.4229 0.5255 0.2947 0.063 0.1078 rs9322817 0.1088 0.486 0.5335 0.1997 0.0924 0.1997 rs2983521 0.2401 0.2857 0.5563 0.9692 0.8259 0.8154 rs10486365 0.6508 0.7737 0.4753 0.5017 0.4047 0.5013 rs6977660 0.9095 0.8635 0.0868 0.1208 0.0691 0.254 rs10499559 0.2781 0.2987 0.8155 0.4067 0.9108 0.6107 rs10486653 0.4469 0.6107 0.6649 0.8594 0.0976 0.1143 rs1527680 0.3504 0.4786 0.2706 0.2082 0.0275 0.0453 rs2252696 0.906 0.8397 0.6661 0.371 0.5509 0.2777 rs7865184 0.5114 0.3136 0.6618 0.7817 0.2243 0.0353 rs10512065 0.8573 0.9703 0.0316 0.0055 0.9892 0.9895 rs965513 0.0052 0.048 0.1351 0.4406 0.143 0.2477 rs1443433 0.2695 0.1495 0.4106 0.216 0.9807 0.7001 rs1443432 0.079 0.0707 0.5559 0.9126 0.3595 0.3215 rs894673 0.1014 0.3248 0.8757 0.8254 0.9111 0.9866 rs3021523 0.3693 0.4695 0.1325 0.2279 0.2819 0.424 rs4460498 0.0998 0.3012 0.8457 0.8253 0.8716 0.9881 76 Table 3-4 Continued rs4128956 0.1921 0.2677 0.3602 0.489 0.4252 0.4105 rs657152 0.119 0.2615 0.3384 0.5335 0.177 0.3247 rs944289 0.9647 0.9142 0.1661 0.1728 0.739 0.4829 rs10149689 0.6685 0.515 0.5691 0.5821 0.3845 0.6665 rs4903957 0.542 0.2547 0.6337 0.4733 0.4111 0.2773 rs11159483 0.7336 0.509 0.1675 0.0578 0.7113 0.9163 rs2075179 0.7537 0.6832 0.9833 0.782 0.8505 0.9743 rs12885526 0.2137 0.1706 0.8077 0.2871 0.853 0.7218 rs1991517 0.4723 0.5476 0.0356 0.258 0.3726 0.517 rs945006 0.4875 0.252 0.6699 0.6907 0.4003 0.4448 rs3908399 0.0187 0.1441 0.6776 0.8059 0.3069 0.085 rs6030171 0.6954 0.6168 0.9116 0.8281 0.3811 0.7481 rs500243 0.3088 0.0431 0.462 0.1649 0.6851 0.5981 rs4892386 0.1641 0.0067 0.0346 0.0094 0.6943 0.8527 Table 3-5 TSH means and standard deviations for PDE8B and FOXE1 significant SNPs. Gene SNP PDE8B rs4704397 FOXE1 SHL term (N=799) SHL preterm (N=404) NICU preterm (N=380) All Cohorts Mean(SD) P-value Mean(SD) Mean(SD) P-value P-value 0.04 1.3x10-4 0.14 6.4x10-4 0.36 0.02 0.28 0.03 0.02 P-value 0.06 AA 10.2±4.5 8.0±4.6 6.2±3.9 GA 8.7±4.6 7.5±4.6 6.2±3.9 GG 8.4±4.3 6.7±4.3 5.3±3.5 -3 rs965513 5.2x10 0.14 AA 9.0±5.0 6.6±3.9 5.1±3.4 GA 8.5±4.3 7.1±4.3 5.8±3.7 GG 9.3±4.6 7.7±4.8 6.2±3.9 rs1443432 0.08 0.56 AA 9.3±4.5 7.6±4.8 6.1±3.9 AG 8.6±4.4 7.3±4.5 5.9±3.8 GG 8.8±4.9 7.1±3.8 5.1±3.2 rs3021523 0.37 0.13 CC 8.5±4.4 6.8±3.2 5.6±2.5 TC 8.7±4.4 6.9±4.6 5.7±3.5 TT 9.1±4.6 7.6±4.6 6.1±4.0 77 Table 3-6 TSH means and standard deviations for marginally significant SNPs. Gene SNP Chr 2 rs9308765 ABO SLC16A2 SHL term (N=799) SHL preterm (N=404) NICU preterm (N=380) All Cohorts Mean(SD) Mean(SD) Mean(SD) P-value P-value 0.20 0.01 0.18 0.02 0.69 0.01 0.03 0.04 P-value 0.04 P-value 0.25 AA 6.7±2.9 8.5±4.1 5.1±3.4 GA 9.5±5.1 7.6±4.6 6.7±4.3 GG 8.8±4.4 7.3±4.5 5.7±3.6 rs657152 0.12 0.34 GG 8.6±4.2 7.0±4.2 5.6±3.4 GT 9.0±4.6 7.5±4.6 5.9±3.8 TT 9.8±5.3 8.0±5.2 6.9±4.7 rs4892386 0.16 0.03 CC 8.8±4.5 6.8±4.2 6.0±3.9 TC 8.1±3.9 8.7±5.0 5.5±3.2 TT 9.8±4.7 7.9±4.7 5.7±3.7 PPP1R9A rs1527680 0.35 0.27 AA 8.9±4.6 7.1±4.2 5.7±3.7 GA 9.0±4.3 8.3±5.1 6.2±3.6 GG 9.6±4.5 7.3±5.6 8.9±6.0 78 79 Table 3-7 SNP haplotypes in the FOXE1 gene that are significantly associated (p<0.001) with natural log transformed TSH levels. SNPs Gene Haplotype Frequency Beta P-value FOXE1 rs965513 - rs1443433 AT 0.23 -0.08 4.2x10-4 GT 0.62 0.07 4.6x10-4 Frequency: frequency of haplotype indicated. Beta: beta coefficient for linear regression model; positive value indicates the haplotype is associated with an increase in TSH level, negative value indicates the haplotype is associated with a decrease in TSH level. P-value = p-value for association between natural log transformed TSH and a specific haplotype composed of the alleles listed. 80 Table 3-8 SNP haplotypes in the TSHR gene that are significantly associated (p<0.001) with natural log transformed TSH levels. SNPs Gene Haplotype TSHR rs10149689-rs4903957-rs11159483 AGT TSHR 0.06 -0.16 7.9x10-4 0.05 -0.16 6.2x10-4 0.03 -0.28 4.2x10-5 0.05 -0.17 7.8x10-4 rs10149689-rs4903957-rs11159483-rs2075179 AGTT TSHR P-value rs4903957-rs11159483-rs2075179 GTT TSHR Frequency Beta rs4903957-rs11159483-rs2075179-rs12885526 GTTA Frequency: frequency of haplotype indicated. Beta: beta coefficient for linear regression model; positive value indicates the haplotype is associated with an increase in TSH level, negative value indicates the haplotype is associated with a decrease in TSH level. P-value = p-value for association between natural log transformed TSH and a specific haplotype composed of the alleles listed. 81 Figure 3-1 Association statistics for all genotyped SNPs with neonatal TSH level for combined populations. The X-axis is a list of all genotyped markers. The Y-axis is the log10 of the p-value from the ANOVA analysis. The horizontal dashed lines represent the p-value cutoffs; * p-value = 0.05, ** p-value = 0.001. The solid line is for all populations combined. The dashed line is for the SHL preterm and SHL term combined. The dotted line is for the SHL preterm and the NICU preterm combined. 82 Figure 3-2 Association statistics for all genotyped SNPs with neonatal TSH level for individual populations. The X-axis is a list of all genotyped markers. The Y-axis is the log10 of the p-value from the ANOVA analysis. The horizontal dashed lines represent the p-value cutoffs; * p-value = 0.05, ** p-value = 0.001. The solid line is for the SHL term population. The dashed line is for the SHL preterm population. The dotted line is for the NICU preterm population. 83 CHAPTER 4 CHARACTERIZATION OF FOXE1 AND PDE8B ENHANCER REGIONS FOR A ROLE IN NORMAL VARIATION IN NEONATAL TSH LEVELS The following manuscript is being prepared for submission. Additional authors on this manuscript include Jeffrey Murray, and Kelli Ryckman. In this study, we followed up on the two most significantly observed associations with thyroid stimulating hormone (TSH) levels in newborns (Chapter 3). We sought to identify the etiologic variants that may be responsible for the observed associations by fine mapping adjacent enhancer elements. This was done through sequencing of a small population of term neonates followed by replication of findings in a larger population of term neonates. We adapted the extreme phenotype sampling approach by selecting infants with newborn screening TSH levels that are on either extreme of the TSH distribution (<2.2 – 25.0 µIU/ml) to increase the power of identifying the rare causal variants. Drs. Jeffrey Murray and Kelli Ryckman provided oversight for all aspects of the project and provided input into study design. I conducted all the experiments and analysis reported in this chapter. I also wrote the manuscript. Abstract Background: Thyroid stimulating hormone (TSH) levels are highly variable and largely controlled by genetic factors. Several genome-wide association studies (GWAS) have identified variants associated with TSH levels; however, the causative variants responsible for GWAS signals are unknown. Methods: A population of term Iowa neonates (n=58) with TSH levels at both ends of the normal distribution (<2.2 – 25.0 µIU/ml) were sequenced in six potential enhancer regions for two of the genes most associated with TSH levels; FOXE1 and PDE8B. Identified variants were genotyped in 84 an additional population of 306 term newborns that were also at the two extreme ends of the TSH level distribution. Results: We identified 12 common and three rare variants in sequencing; and although none of these variants were statistically significant with TSH levels, one of the rare variants (rs112053411) identified was interestingly found in three neonates, all in the upper distribution of TSH levels including one infant that was later diagnosed with congenital hypothyroidism. Conclusions: Variants identified were not statistically significant for an association with TSH levels; however, one of the rare variants (rs112053411) identified only in neonates in the upper distribution of TSH levels is worth pursuing with functional studies as well as further replication in individuals with thyroid disorders. Introduction In healthy adult subjects, thyroid stimulating hormone (TSH) has considerable variability between individuals, whereas this variability is much less in the same individual when TSH is measured repeatedly over an extended period of time (Panicker et al. 2010). Likewise, newborn TSH levels are highly variable and this variability is largely controlled by genetic factors (Alul et al. 2013a; Alul et al. 2013b). In the previous work reported in chapters two and three, we have estimated the heritability of TSH levels in newborns to be 58% which mirrors previous heritability estimates for adult serum TSH levels (Panicker et al. 2008), and we identified genetic variants that are associated with neonatal TSH levels. The identified variants (rs965513 upstream of FOXE1 and rs4704397 in intron 1 of PDE8B) replicated previous genome-wide association studies (GWAS) for adult TSH levels; however, the causative variants responsible for the reported associations have not yet been identified. These two variants; rs965513 and rs4704397, have been extensively studied and found to be significantly associated with various thyroid related traits in various studies, hence, pursuing the causative variants responsible for these associations is justified. As 85 for rs965513 which was found to be associated with adult TSH level in GWAS (Gudmundsson et al. 2012), it has been repeatedly found to be associated with thyroid cancer (Gudmundsson et al. 2009; Gudmundsson et al. 2012; Jones et al. 2012; Tomaz et al. 2012) and with hypothyroidism (Denny et al. 2011). Whereas rs4704397 that was initially identified in the GWAS by Arnaud-Lopez et al. (2008) to be associated with adult serum TSH levels, was later reported to be associated with subclinical hypothyroidism during pregnancy (Shields et al. 2009), as well as with TSH levels in obese children (Grandone et al. 2012). Identifying the causative variant responsible for these associations is important to better understand the etiology of these thyroid conditions. In this study, we sequenced potential enhancer regions for the two most significant genes from the previous association study; FOXE1 and PDE8B, to examine possible causative variants. We chose the extreme phenotype sampling approach for study population selection. The extreme phenotype approach has been used to search for rare causal variants in complex traits by selecting individuals at both extremes of a continuous trait distribution spectrum. The principle is that rare causal variants with large effect sizes on the phenotype of interest will be enriched in one of the two tails of the distribution leading to an increased minor allele frequency in this particular study group and therefore an increased power in identifying the rare causal variants compared to random sampling from the entire distribution (Lamina 2011; Emond et al. 2012; Barnett et al. 2013). We also chose a unique approach for the selection of potential enhancer regions of interest by utilizing the recently published data by the Encyclopedia of DNA Elements (ENCODE) (Dunham et al. 2012). The ENCODE project was designed to comprehensively describe all functional elements of the human genome. The most striking finding of the ENCODE project was that about 80% of the human genome is functional and mostly comprises regulatory elements such as enhancers (Dunham et al. 2012). Enhancer elements are critical for the regulation of gene expression and authors 86 from the ENCODE project found that previously reported GWAS single nucleotide polymorphisms (SNPs) are enriched in the identified regulatory elements in this project including enhancers (Dunham et al. 2012). ENCODE findings have a great impact on biomedical research, shed light on the importance of enhancer elements, and provide new insight for the search of causative variants after association studies. In this study, we sequenced potential enhancer regions based on ENCODE data, in a group of 58 term newborns that are at the two extreme ends of the TSH level distribution (<10th and >90th percentile). We identified three rare variants in sequencing results. To follow up on the identified rare variants, we used the TaqMan chemistry to genotype them in an additional population of 306 term newborns that are also at the two extreme ends of the TSH level distribution (<5th and >95th percentile). Materials and Methods This study was conducted in two phases. The first phase is the sequencing of potential FOXE1 and PDE8B enhancer regions in a small population of 58 term neonates. The second phase pursues sequencing results of interest in a larger population of 306 term neonates by using the TaqMan genotyping system (Applied Biosystems, Foster City, CA). Study Population For this study, we used the extreme phenotype sampling approach by selecting infants with newborn screening thyroid stimulating hormone (TSH) levels that are on either extreme of the TSH distribution (<2.2 – 25.0 µIU/ml). Study samples were obtained from two sources. The first source was from the University of Iowa Hospitals and Clinics Newborn Nursery. Only singleton term neonates (n=58) with gestational ages between 37 and 42 weeks were included in this study. Infants with TSH levels below the 10th (n=30) or above the 90th (n=28) percentile of the TSH distribution were chosen. Only 1 individual had an abnormal (TSH≥25.0 µIU/ml) level on the initial screen. Gestational 87 age was determined from the first day of the last menstrual period. Gestational age was confirmed through obstetrical judgment and if uncertain, ultrasound measurements were used for confirmation. All families provided signed informed consent (UI IRB199911068) to be included in a repository of samples designed to provide DNA and limited epidemiologic data to study complications of newborn infants (Steffen et al. 2007). TSH measured by the State Hygienic Laboratory (SHL) at the University of Iowa as part of routine neonatal screening was linked to the sample and medical record data. DNA was extracted from cord blood, discarded venous blood, or buccal swab for all infants. All DNA was extracted using standard protocols. The second source was from the University of Iowa SHL where we obtained deidentified residual dried blood spots from a population of Iowa newborns as part of the newborn screening program. Only singleton term neonates (n=306) with gestational ages between 39 and 41 weeks were included in this study. Infants with TSH levels below the 5th (n=154) or above the 95th (n=152) percentile of the TSH distribution were selected. Approval for use of the de-identified data and blood spot cards was granted by the Iowa Department of Public Health and a waiver of consent was obtained from the Institutional Review Board at the University of Iowa (IRB#200908793). All neonates had TSH measurements obtained between day 1 and 3 of life and had not received a transfusion prior to sample collection. All neonates with a presumptive positive screening result were excluded from this study. Quantification of TSH was performed at the SHL using CLIA (Clinical Laboratory Improvement Amendments) certified methods. TSH was determined by solid phase, time-resolved fluoroimmunoassay from dried newborn blood spots using PerkinElmer’s AutoDELFIA platform (Waltham, MA). DNA was extracted from one dried whole blood spot for all samples using the AutoGen (Holliston, MA) QuickGene810 nucleic acid extraction machine with the DNA Tissue Kit (AutoGen) and following manufacturer’s recommendations. 88 Selection of Studied Regions We selected six potential enhancer regions for FOXE1 and PDE8B genes based on ENCODE data (Table 4-1) using the University of California, Santa Cruz (UCSC) Genome Browser (Kent et al. 2002; Rosenbloom et al. 2013). The specific UCSC Genome Browser ENCODE tracks used were the “Layered H3K27Ac” track which is for an activating acetylation mark on seven cell lines from ENCODE, one of the histone modification marks often found near active regulatory elements and believed to differentiate between active and inactive enhancers, and the “DNase Clusters” track which is for DNaseI hypersensitive sites in 125 cell types from ENCODE, marking accessible chromatin which is often found in regulatory DNA regions (Dunham et al. 2012). The mammalian, primate, and vertebrate conservation tracks form UCSC Genome Browser were also used to see if the picked regions were conserved (Figure 4-1 and Figure 4-2). Potential enhancer regions were chosen in the vicinity of the two most significantly associated SNPs with neonatal TSH levels in the work reported in Chapter three (rs965513 near FOXE1, and rs4704397 near PDE8B) (Figure 4-1 and Figure 4-2). These two SNPs are also among the top GWAS associated SNPs with adult TSH levels and adult thyroid disorders (Arnaud-Lopez et al. 2008; Gudmundsson et al. 2009; Shields et al. 2009; Denny et al. 2011; Grandone et al. 2012; Gudmundsson et al. 2012; Jones et al. 2012; Tomaz et al. 2012). Genotyping Sequencing Primers used to amplify all enhancer regions were designed with Primer 3. Primer sequences and PCR conditions for amplifying the regions of interest for the sequencing of enhancer regions are listed in Table 4-2. Amplified regions in the form of PCR products were sent for sequencing using an ABI 3730XL (Functional Biosciences, Inc., Madison, WI). The six potential FOXE1 and PDE8B enhancer regions were sequenced in 89 58 term neonates in the forward direction. All identified rare variants were confirmed by sequencing in the reverse direction. Chromatograms were transferred to a UNIX workstation, base-called with PHRED (v.0.961028), assembled with PHRAP (v.0.960731), scanned by POLYPHRED (v.0.970312), and viewed with the CONSED program (v.4.0). TaqMan We selected three SNPs to follow up on with TaqMan genotyping based on sequencing results (Table 4-3). We chose to follow up on all three rare SNPs, minor allele frequency (MAF) < 1%, identified in sequencing, and none of the common SNPs since they were all not significantly associated. SNPs were genotyped using the TaqMan chemistry (Applied Biosystems, Foster City, CA). All SNP genotyping assays were available and ordered using either the Assay-on-Demand or Assays-by-Design service from Applied Biosystems. These SNP genotyping assays included primers to amplify the region containing the SNP of interest and two TaqMan Minor Groove Binder probes that are specific to the polymorphic variant alleles at the site labeled with different fluorescent reporter dyes, FAM and VIC. PCR reactions were performed using standard conditions supplied by Applied Biosystems. Following thermocycling, fluorescence levels of the FAM and VIC dyes were measured using the Applied Biosystems 7900HT Fast System and genotypes were scored using the Sequence Detection System (SDS) software (Applied Biosystems). Genotypes were uploaded into a Progeny database (Progeny Software, LLC, South Bend, IN) containing the phenotypic data for subsequent analysis. Genotyping efficiency was >98% for all markers. Statistics Given that we used the extreme phenotype approach for study population selection, we treated TSH levels as a dichotomous trait by grouping all sequenced neonates with low TSH values (n=30) into one group and all sequenced neonates with 90 high TSH values into another group (n=28). To determine if any of the observed sequence variants is associated with newborn TSH levels, we compared the frequency of each sequence variant between the low TSH group and the high TSH group with Fisher’s exact test. We excluded any sample with poor sequencing quality (PHRED Quality Score <30) for a given variant from analysis for that specific variant. For the three SNPs genotyped using TaqMan assays, testing for association of with TSH levels in a group of 306 neonates was performed as mentioned above. Results Sequencing A total of 58 infants were sequenced for six FOXE1 and PDE8B enhancer regions to search for potential etiologic sequence variants that may be associated with newborn TSH levels. A total of 15 known sequence variants were detected; 14 of which are SNPs and one deletion/insertion variation (DIV) of 8 base pairs (Table 4-4). Of the 15 identified sequence variants, 12 are common (MAF > 1%), were not found to be associated with newborn TSH levels (p-value > 0.05), and will not be pursued any further. The remaining 3 SNPs are rare (reported MAF < 1% in the 1000 genomes project) and each SNP was identified in one sample only. SNPs rs148044470 in PDE8B Enhancer 1 and rs117811946 in FOXE1 enhancer region 1 were each found in an infant with TSH level below the 10th percentile. SNP rs112053411 in FOXE1 enhancer region 3 was found in an infant with TSH level above the 90th percentile. The TSH level for this infant was interpreted as borderline in the newborn screen for congenital hypothyroidism (CH). After sequencing and identifying this SNP, we searched the medical record and found that this infant was later diagnosed with CH based on follow-up serum testing in the clinic. We then sequenced this infant’s parents and found that this SNP is transmitted from the mother who is also heterozygous for it and was diagnosed with hypothyroidism. 91 TaqMan A total of 306 infants were genotyped with TaqMan assays for the three rare SNPs identified in sequencing (rs148044470, rs117811946, and rs112053411) to increase the power of detecting an association with TSH levels. SNP rs148044470 was identified in two samples, one from the low TSH group and one from the high TSH group. SNP rs117811946 was identified in five samples, two from the low TSH group and three from the high TSH group. SNP rs112053411 was identified in two samples, both from the high TSH group. All three SNPs were not found to be associated with newborn TSH levels (pvalues 1.0, 0.68, and 0.50 respectively). Discussion The genetic contribution to TSH variability is widely studied due to the importance of TSH level in normal human physiology. There have been six successful GWAS, to date, identifying a number of loci to be associated with TSH variability (Hwang et al. 2007; Arnaud-Lopez et al. 2008; Gudmundsson et al. 2009; Lowe et al. 2009; Panicker et al. 2010; Gudmundsson et al. 2012). One of the most interesting associated loci is a region in intron 1 of PDE8B, which has been found to be the most significantly associated locus (p-value=1.95x10-56) in the most recent meta-analysis of TSH level related GWAS (Porcu et al. 2013). An attempt to identify the functional variant responsible for this association by sequencing all PDE8B exons was unsuccessful, suggesting that the causative variant is likely in a noncoding region (Arnaud-Lopez et al. 2008). A second interesting locus is an intergenic region on chromosome 9q22, with FOXE1 being the nearest gene, found in a GWAS of TSH level to be one of the most significantly associated loci (p-value=4.3x10-31) (Gudmundsson et al. 2012). The causative variant responsible for this association has not been identified either since most GWAS identify associated regions but not the causative variants. It has been observed that about 40% of GWAS significantly associated SNPs are located in intergenic regions, 92 and another 40% are in introns (Manolio 2010). Furthermore, research from the ENCODE project has shown that previously reported GWAS SNPs are enriched in the identified regulatory elements in this project including enhancers (Dunham et al. 2012). Examples of diseases known to be caused by enhancer variants include α-thalassemia, preaxial polydactyly, holoprosencephaly, and nonsyndromic cleft lip (Epstein 2009). Therefore, comparing GWAS associated loci for a given trait, such as TSH levels, with reported ENCODE regulatory regions may help identify narrower loci that are functional and worth pursuing with fine mapping techniques. Fine mapping a given region to identify causative variants may be done through sequencing which enables the identification of rare variants that are missed in GWAS and may have a more extreme phenotype. In this light, we decided to further investigate these two loci (PDE8B and FOXE1), which were also the most significantly associated with newborn TSH level in the work previously reported in Chapter 3. We aimed to fine map six potential enhancer regions identified by ENCODE that were also in the vicinity of the above mentioned GWAS hits to identify potential causative variants. Only term newborns were selected to minimize TSH variability due to gestational age and birth weight factors. We identified a total of 15 known variants in sequencing the first population of 58 newborns. Of the 15 identified variants, 12 are common (MAF > 1%) and were not found to be associated with newborn TSH levels (p-value > 0.05). The remaining 3 SNPs are rare (MAF < 1%) and each SNP was identified in one sample only. Our most interesting finding is SNP rs112053411 in the FOXE1 enhancer region 3 identified in an infant with an initial TSH level above the 90th percentile. The TSH level for this infant was interpreted as borderline in the newborn screen for CH. However, after identifying this SNP through sequencing, we examined the medical record for this infant and found that the infant was later diagnosed with CH based on follow-up serum testing in the clinic. We also discovered that the infant’s mother is on thyroid hormone 93 replacement therapy. After sequencing this infant’s parents, we found that the mother is also heterozygous for this SNP. Whether the mother had CH or adult-onset hypothyroidism is not known, however, the fact that both mother and infant are hypothyroid and heterozygous for this rare SNP is intriguing. When we further examined this SNP with TaqMan genotyping, it was identified in two infants from the high TSH group (n=152) and in none of the infants in the low TSH group (n=154). Although this finding was not statistically significant for an association with TSH levels (p-value = 0.50), this may be due to the modest study population size and lack of power to detect an association for SNPs with small effect sizes. Furthermore, given that this SNP is very rare with MAF of 4 in a thousand, and yet it was detected in a total of two infants in TaqMan genotyping, all with high TSH levels, from a small population size of 306, it can be argued that this SNP is worth pursuing further in a larger population. Although the observed MAF for this SNP in our population of 306 infants is not statistically significantly different from the reference MAF (p-value = 0.63), it is still observed almost two times more than expected making it an interesting finding. If this SNP shows statistical significance for association with TSH levels in future larger studies, or is shown to affect enhancer activity in functional studies, then this SNP might have important clinical implications. Especially since there has been recent evidence that infants with elevated TSH in newborn screening (blood spot TSH > 20 µIU/ml), but are not diagnosed with CH, are at an increased risk of developing persistent subclinical hypothyroidism later in childhood (Leonardi et al. 2008). Identifying infants who are likely to develop thyroid problems later in childhood is critical for early detection and treatment. The two other rare SNPs identified are rs148044470 in PDE8B Enhancer 1 and rs117811946 in FOXE1 enhancer region 1, each found in an infant with TSH level below the 10th percentile. However, after doing TaqMan genotyping of 306 infants, the two 94 SNPs were found in infants from both the low and the high TSH groups, and therefore are unlikely to be associated with TSH levels (p-values 1.0 and 0.68 respectively). This study has a few limitations; first, we do not have access to the medical records for SHL studied infants since we only obtained their de-identified data and blood spot cards. Therefore, we were not able to connect TSH measurements to medical record information to obtain more detailed information on neonatal illness or race. However, in 2009 86.9% of the births in Iowa were Caucasian, suggesting that the majority of our samples are from Caucasians. Another limitation was that we do not have follow up data on our study cohort to check for development of thyroid conditions later in life. As mentioned above, studies have shown a high rate of persistent subclinical hypothyroidism developing later in childhood for infants with elevated TSH in newborn screening (blood spot TSH > 20 µIU/ml), but are not diagnosed with CH (Leonardi et al. 2008). Having that information would have been valuable to check for an association with our studied SNPs. And finally, our small study size does not give us enough power to detect variants with low effect sizes for a trait such as TSH level which is a highly polygenic and complex trait. 95 Table 4-1 Potential FOXE1 and PDE8B enhancer regions studied. Region Location (UCSC Genome Browser February 2009 build) Distance from top GWAS associated SNP FOXE1 Enhancer 1 chr9:100,564,149-100,565,129 8 kb downstream of rs965513 FOXE1 Enhancer 2 chr9:100,565,744-100,566,698 10 kb downstream of rs965513 FOXE1 Enhancer 3 chr9:100,535,465-100,536,666 19 kb upstream of rs965513 PDE8B Enhancer 1 chr5:76,587,369-76,588,272 69 kb downstream of rs4704397 PDE8B Enhancer 2 chr5:76,541,540-76,541,997 23 kb downstream of rs4704397 PDE8B Enhancer 3 chr5:76,597,139-76,598,388 79 kb downstream of rs4704397 Source: (UCSC Genome Browser). 96 Table 4-2 Primer sequences and PCR conditions for sequencing FOXE1 and PDE8B enhancer regions. Amplicon Primer Sequence Annealing Temperature Product Size FOXE1 Enhancer 1 F R CGTTCTTCCTTTTCACTTGCT TCTGTTCTTTTTCCCTTTTGG 55 843 FOXE1 Enhancer 2 F R GACAGGTGGCCTCGCATC TGTCCCACTCCATCCCTCTC 62 955 FOXE1 Enhancer 3 F1 R1 F2 R2 GGGAAGTATGGTGGGAACCT GCTACAGTTGGGTCTGGAAAC GACATTCTCGCCTTTCACACT TTCCAATAACAGTCCTCCGATT 62 910 56 511 PDE8B Enhancer 1 F R AATCTTTCGCTGGCTTGGAC CACAACATACGCCTTCCTGA 61 867 PDE8B Enhancer 2 F R GCCTGCCTCATTTCCTCTTT GAGTCCCCACGAACAACCTA 61 779 PDE8B Enhancer 3 F1 R1 F2 R2 TCTGTTTCTTTGCCCATCAA GCCATCGTCTTGCTAACTCTG CGGCAGTGTCTATTATCAGTGG CAGGTAGGGAAGTCGGTGTG 61 900 61 945 97 Table 4-3 List of SNPs selected for further investigation after sequencing. SNP Base change Region MAF Reason SNP was chosen rs117811946 C/G FOXE1 Enhancer 1 0.004 SNP was found in sequencing in one neonate with TSH below the 10th percentile rs112053411 C/G FOXE1 Enhancer 3 0.004 SNP was found in sequencing in one neonate with TSH above the 90th percentile that was later diagnosed with congenital hypothyroidism rs148044470 A/T PDE8B Enhancer 1 0.002 SNP was found in sequencing in one neonate with TSH below the 10th percentile Table 4-4 List of all sequence variants detected in the sequencing of 58 newborns. Region Sequence variation name in dbSNP Variation class Change Minor allele frequency (MAF) Location (UCSC Genome Browser February 2009 build) Number of newborns with variant (low and high TSH group) Fisher’s exact test pvalue for association with TSH level FOXE1 Enhancer 1 rs79229991 SNP (A/C) 0.03 chr9:100,564,393 5 (3 and 2) 1.0 rs57077725 SNP (C/T) 0.04 chr9:100,564,512 5 (3 and 2) 1.0 rs117811946 SNP (C/G) 0.004 chr9:100,564,806 1 (1 and 0) 1.0 FOXE1 Enhancer 2 rs77394667 SNP (A/G) 0.13 chr9:100,565,813 18 (11 and 7) 1.0 rs114804130 SNP (C/G) 0.10 chr9:100,566,031 12 (8 and 4) 0.74 FOXE1 Enhancer 3 rs112053411 SNP (C/G) 0.004 chr9:100,536,171 1 (0 and 1) 0.49 rs62573974 SNP (C/T) 0.34 chr9:100,536,542 33 (15 and 18) 0.62 rs141224302 DIV (8bp del) 0.34 chr9:100,536,606100,536,613 33 (15 and 18) 0.62 PDE8B Enhancer 1 rs148044470 SNP (A/T) 0.002 chr5:76,587,716 1 (1 and 0) 1.0 PDE8B Enhancer 2 rs57774448 SNP (G/C) N/A chr5:76,541,674 19 (10 and 9) 1.0 PDE8B Enhancer 3 rs10066802 SNP (A/G) 0.44 chr5:76,597,461 27 (10 and 17) 0.33 rs76281411 SNP (C/T) 0.22 chr5:76,597,627 18 (8 and 10) 0.79 rs11745130 SNP (A/G) 0.16 chr5:76,597,857 24 (13 and 11) 0.81 rs35689253 SNP (A/G) 0.32 chr5:76,597,887 35 (19 and 16) 0.83 rs1382893 SNP (C/T) 0.13 chr5:76,598,292 9 (4 and 5) 1.0 98 Figure 4-1 FOXE1 potential enhancer regions. The GWAS signal rs965513 is shown in green and the three selected enhancer regions are shown with UCSC Genome Browser ENCODE regulation tracks to demonstrate enhancer signals. 99 Figure 4-2 PDE8B potential enhancer regions. The GWAS signal rs4704397 is shown in green and the three selected enhancer regions are shown with UCSC Genome Browser ENCODE regulation tracks to demonstrate enhancer signals. 100 101 CHAPTER 5 CONCLUSIONS AND FUTURE DIRCTIONS Conclusions Congenital hypothyroidism (CH) is defined as thyroid hormone deficiency present at birth. It is a common and preventable cause of mental retardation and is screened for at birth in the newborn screening program by thyroid stimulating hormone (TSH) or thyroxine measurements. TSH is considered the most informative marker of thyroid function and is the test of choice for clinicians for the diagnosis of most thyroid disorders. However, TSH is highly variable and manifests a wide reference range in the general population leading to some challenges in its interpretation in newborn screening as well as its later use as a clinical diagnostic test of thyroid function. This variability in TSH is largely controlled by genetic factors and this has been extensively studied in adults, however, no studies have addressed neonatal TSH variability. The work in this thesis is focused on exploring whether TSH variability is largely controlled by genetic factors in the neonatal period as it is in adults, as well as investigating these genetic factors in both term and preterm neonates. The genetic factors controlling adult TSH variability have been widely studied through heritability studies, candidate-gene studies, and genome-wide association studies (GWAS). GWAS have no a priori hypotheses and therefore have the advantage of discovering loci that would not be suspected to be associated based on known biology. Hence, several novel loci have emerged from GWAS associated with adult TSH levels; however, the causative variants responsible for the reported associations were not identified. This work addresses all of these areas but in relation to neonatal TSH variability rather than adult. We estimated heritability, replicated previous genetic associations reported in GWAS, and investigated functional variants that may be responsible for GWAS signals. 102 In case of TSH heritability; adult TSH heritability was estimated to vary from 32% (Samollow et al. 2004), to 64% (Hansen et al. 2004), and to a maximum of 65% (Panicker et al. 2008). TSH heritability has never been determined in the neonatal period; therefore, we examined 381 twin pairs obtained from the Iowa Neonatal Metabolic Screening Program. We estimated neonatal TSH heritability to be 58% with a P-value of 2x10-5. TSH is known to be largely affected by age, and the use of age-dependent reference ranges for TSH is strongly advocated, with infants, children, adolescents, and adults being in separate groups (Kratzsch et al. 2008). Therefore, it can be argued that different genetic and environmental factors are responsible for this age-dependent variation, resulting in different heritabilities across different age groups. Furthermore, there are examples from the literature where heritability estimates vary between different age groups, such as the variable heritability estimates of lipid and lipoprotein levels across three age groups in the same study (Iliadou et al. 2001). Our study is the first to estimate heritability of TSH levels in the neonatal period, as well as the first to use newborn blood spot card measurements to estimate the genetic heritability of any trait, illustrating the utility of stored newborn blood spot cards as a valuable resource for genetic studies. The estimated heritability of 58% in this study mirrors previous reports of adult TSH heritability, and provides direct evidence for a strong genetic contribution to TSH variability at birth, suggesting that thyroid function is mainly controlled by genes throughout life. This may have clinical implications in utilizing neonatal TSH measurements to predict the risk of adult diseases later in life. For example, TSH level variation even within the normal physiological range in euthyroid adults has been associated with bone mineral density (van der Deure et al. 2008), mean platelet volume possibly affecting thrombosis (Kim et al. 2013b), and cholesterol and triglycerides (Roos et al. 2007; Garduno-Garcia et al. 2010). And since TSH is largely controlled by genetic factors, it can be argued that neonatal TSH levels may reflect TSH 103 levels in adulthood and provide early prediction of childhood and adult thyroid abnormalities. Given that TSH individual levels are highly heritable, efforts were directed towards identifying the genes responsible for this heritability. Several GWAS as well as candidate-gene studies were successful in identifying both specific genes as well as intergenic loci with unknown function to be associated with adult TSH levels. However, no studies addressed the genetic variation associated with neonatal TSH levels, and as mentioned previously different genes may play a role at different stages for a given trait. Therefore, we aimed to replicate major genetic associations with adult TSH levels from the literature in a population of term and preterm neonates, as well as study other candidate genes that are biologically plausible to control TSH levels but were not found to be significantly associated in adult TSH studies. Our results replicated two of the most important adult GWAS findings (Arnaud-Lopez et al. 2008; Gudmundsson et al. 2012), the first was for rs4704397 in the PDE8B gene (p=1.3x10-4), and the second was rs965513 (p=6.4x10-4) on chromosome 9 upstream of the FOXE1 gene. Both of these SNPs met statistical significance after correction for multiple testing. Our results demonstrate the importance of these two loci in determining TSH levels in the newborn period, emphasizing their role at birth and in adulthood. However, thyroid function undergoes marked changes during puberty to adapt to the increased energy expenditure in response to body and sexual development (Fleury et al. 2001). Therefore, additional studies in late childhood and adolescence are needed to ensure their involvement throughout life, and it could be speculated that additional genetic factors may be responsible for the observed thyroid function changes during puberty. Additionally, these two SNPs have been extensively studied and found to be significantly associated with various thyroid related diseases in various studies. For example, rs965513 has been repeatedly found to be associated with thyroid cancer (Gudmundsson et al. 2009; Gudmundsson et al. 2012; Jones et al. 2012; Tomaz et al. 104 2012) and with hypothyroidism (Denny et al. 2011). For thyroid cancer, each copy of the minor A allele for SNP rs965513 near FOXE1 was associated with an increased risk of thyroid cancer (Gudmundsson et al. 2009; Gudmundsson et al. 2012; Jones et al. 2012; Tomaz et al. 2012). The minor A allele is also the allele found to be associated with a decrease of 0.2-0.7 mIU/L in TSH levels in our term and preterm combined cohort, which mirrors adult findings. It has been speculated that lower TSH levels may lead to poor differentiation of thyroid epithelium, resulting in an increased risk of malignant transformation (Gudmundsson et al. 2012). As for rs4704397, it was reported to be associated with subclinical hypothyroidism during pregnancy (Shields et al. 2009), as well as with TSH levels in obese children (Grandone et al. 2012). Therefore, the identified association of these two SNPs with TSH levels at birth sheds light on the possibility of using these two SNPs as biomarkers for the early prediction of risk for adult diseases and conditions associated with thyroid hormone levels. Finally, we aimed to identify the causative variants that may be responsible for the observed associations with rs4704397 in intron 1 of the PDE8B gene and rs965513 in an intergenic region on chromosome 9 upstream of the FOXE1 gene. Both these variants have been identified in adult TSH GWAS (Arnaud-Lopez et al. 2008; Gudmundsson et al. 2012) but the functional variants responsible for these associations are unknown. Identifying the real functional variants tagged by the associated SNP after GWAS has always been a challenge. Often, functional variants are unsuccessfully searched for in the nearest gene. This is the case with rs4704397 in intron 1 of the PDE8B gene where all exons were sequenced but the causative variant was not identified (Arnaud-Lopez et al. 2008). This implies that the causative variant may be in intergenic regions that function as regulatory elements controlling gene expression, such as enhancers. The search for etiologic variants in noncoding regulatory regions can be argued for especially after the Encyclopedia of DNA Elements (ENCODE) project reported that GWAS significantly associated SNPs are enriched in the identified regulatory elements in this project 105 including enhancers (Dunham et al. 2012). We chose to fine map six enhancer regions for our two genes of interest PDE8B and FOXE1 to identify functional variants that may be affecting enhancer activity resulting in differential gene expression and variable TSH levels. None of the identified variants were statistically significant for an association with TSH levels; however, one of the rare variants was only identified in neonates with TSH levels above the 95th percentile of the distribution, one of whom was diagnosed with CH, making this variant worth pursuing with functional studies. There are several challenges to identifying genetic factors responsible for the variation observed in a complex trait such as TSH level. First, although there have been several successful GWAS and candidate-gene studies identifying variants associated with adult TSH levels, all identified loci together do not account for the entire observed variability in TSH levels. For example, it is reported that the most significantly associated SNP with TSH levels in PDE8B gene (rs4704397) is responsible for 2.3% of the observed variation in TSH levels (Arnaud-Lopez et al. 2008). Furthermore, the most recent meta-analysis of GWAS for TSH variation reported that all 19 significantly associated loci account for only 5.64% of the total trait variance observed (Porcu et al. 2013).Therefore, it is evident that there are more genes and variants yet to be identified to explain the remaining missing heritability. It has been suggested that missing heritabilities of complex traits may be within a large number of unidentified variants each with a small effect size making their identification hard in small study cohorts (Manolio et al. 2009). Another possibility is the role of rare variants, each with larger effect sizes, however also difficult to detect in traditional GWAS and candidate-gene association studies (Manolio et al. 2009). And finally, missing heritabilities may be explained by structural variants, gene-gene, or gene-environment interactions that are poorly studied with current technology (Manolio et al. 2009). A second challenge relates to understanding the biology and pathways responsible for the observed associations. For example, the FOXE1 SNP rs965513 that was 106 repeatedly found to be associated with an increased risk of thyroid cancer (Gudmundsson et al. 2009; Gudmundsson et al. 2012; Jones et al. 2012; Tomaz et al. 2012) and the same risk allele is associated with low TSH levels in adults and neonates. This risk allele has a frequency of 37% leading to the fundamental question of why this risk allele was not influenced by negative selection. There might be underlying beneficial traits that are associated with this risk allele resulting in the observed high frequency. In this case, studies have found that high TSH levels within the normal range are a risk factor for adult thyroid cancer (Kim et al. 2013a) as well as pediatric thyroid cancer (Chiu et al. 2012). This may explain the persistence of the risk allele resulting in low TSH level, but at the same time does not explain how the same allele is associated with an increased risk of thyroid cancer. This leads to the recognized conclusion that the exact mechanisms involved are yet to be identified. A third challenge in studying the genetic factors affecting neonatal TSH levels is the large influence of gestational age, birth weight, and infant sickness on neonatal TSH levels. This explains the less significant associations that we observed in our preterm population compared to the term population when we studied genetic factors affecting TSH levels in neonates (Alul et al. 2013b). There is also a challenge related to the use of de-identified samples such as the study population that we obtained from the State Hygienic Lab (SHL). Given that the samples are de-identified, we had no information on infant sickness to account for when doing the association study with TSH levels. Furthermore, we did not have diagnostic information on SHL infants with borderline TSH levels in newborn screening and we could not determine if any of them were later diagnosed with CH, therefore we had to exclude all SHL infants with borderline TSH levels from our study although this borderline population may be the best sample to examine for rare variants with large effect sizes. 107 Future Directions In iodine sufficient countries, abnormal thyroid gland development is the most common cause of congenital hypothyroidism (CH) accounting for 80-85% of CH cases (Hinton et al. 2010; Rastogi and LaFranchi 2010). Thyroid dysgenesis CH is mostly sporadic; only 2% of cases have a known causing mutation in one of the genes necessary for thyroid development (NKX2.1, NKX2.5, PAX8, or FOXE1), whereas the remaining 98% of cases have an unknown cause (Rastogi and LaFranchi 2010; Abduljabbar and Afifi 2012). So, it is evident that we still have much to learn about the genetic etiology of CH. In thyroid dysgenesis CH cases with a mutation in one of the genes encoding a transcription factor, CH may be present as part of a syndrome; for example FOXE1 mutations lead to Bamforth-Lazarus Syndrome (Rastogi and LaFranchi 2010). So, it may be hypothesized that in the remaining sporadic non-syndromic thyroid dysgenesis CH cases, the presence of a genetic variant within an enhancer element may be the cause of CH. This hypothesis can be supported by the fact that enhancer elements are often tissue specific (Epstein 2009). So a genetic variant affecting thyroid specific enhancer activity may lead to thyroid dysgenesis but not affect gene expression in other tissues therefore leading to non-syndromic CH. The involvement of genetic variants as the underlying cause of a sporadic disease may be supported with the argument for gene-gene and geneenvironment interactions. Moreover, sporadic CH is not entirely sporadic, it has been shown that there is approximately a 2% CH recurrence risk in families of newborns with thyroid dysgenesis (Rastogi and LaFranchi 2010). Further evidence of genetic involvement in what is thought to be a sporadic occurrence was shown in a French study reporting that for patients with thyroid dysgenesis CH, 21.4% of first degree relatives had asymptomatic thyroid developmental anomalies, compared to 0.9% in controls leading to the hypothesis of a common genetic component with heterogeneous phenotypes (Leger et al. 2002). This is not unusual in polygenic or complex disorders and may even be observed in Mendelian disorders such as the report of a monozygotic twin pair carrying 108 the same mutation for Van der Woude Syndrome, which has an autosomal dominant inheritance pattern, but are discordant for the phenotype (Jobling et al. 2011). The authors suggest that this discordance may be explained by genetic and epigenetic factors as well as the intrauterine environment (Jobling et al. 2011). To study potential enhancer involvement in CH etiology we recently submitted a proposal for review by the University of Iowa Institutional Review Board (IRB) to recruit children diagnosed with CH from the University of Iowa Hospitals and Clinics pediatric endocrinology clinic. We plan to recruit children diagnosed with CH, their siblings, parents, and potentially grandparents, uncles, aunts, or cousins if any have thyroid problems. The aim of this study would be to examine potential enhancer elements for any genetic variants that may be associated with the disease. Identifying the underlying genetic cause is of great importance for genetic counseling of the family for future pregnancies. Although, as mentioned above, the recurrence risk is only about 2% for “sporadic” thyroid dysgenesis (Rastogi and LaFranchi 2010), there is an increased prevalence of asymptomatic thyroid developmental anomalies (21.4% versus 0.9% in controls (Leger et al. 2002)) and these asymptomatic relatives may pass the CH risk to their offspring. Additionally, one proof of enhancer activity may be done in vivo through a transgenic reporter assay. In the work reported in chapter four we selected enhancer regions for fine mapping based on ENCODE data. One of the SNPs (rs112053411 in FOXE1 enhancer region 3) identified is especially interesting and worth pursuing; it is a rare variant that was only identified in neonates with TSH levels above the 95th percentile of the distribution, one of whom was diagnosed with CH. We believe that this enhancer element (FOXE1 enhancer region 3) should be studied further in a thyroid cell line to confirm enhancer activity in thyroid tissue. Studying the effect of the identified SNP (rs112053411) on enhancer activity and FOXE1 gene expression is also necessary. If it is proved to be an active enhancer for FOXE1 gene expression in thyroid cell lines, then 109 additional studies of the enhancer activity with the common and rare alleles of SNP rs112053411 in a mouse model would be necessary to evaluate effect on thyroid embryogenesis. The work reported in this thesis explored the contribution of genetic variants to the wide range of normal variation observed in TSH levels in newborns. As a result, we estimated this genetic contribution to TSH level variation in newborns for the first time to be 58%. We identified two genetic variants to be significantly associated with newborn TSH level variation, replicating previous findings in adults. And finally, the work reported here sheds light on the importance of fine mapping enhancer elements in studying the genetic contribution to complex traits. In the future, the use of larger study cohorts combined with detailed phenotyping especially of relatives of CH patients with asymptomatic thyroid anomalies will give insights into the true etiology of CH. Ultimately, this work can have important clinical implications for better screening, diagnosis, and counseling of CH families for future pregnancies. 110 REFERENCES Abduljabbar, M. A. and A. M. Afifi (2012). "Congenital hypothyroidism." Journal of pediatric endocrinology & metabolism : JPEM 25(1-2): 13-29. Almandoz, J. P. and H. Gharib (2012). "Hypothyroidism: etiology, diagnosis, and management." The Medical clinics of North America 96(2): 203-221. Altshuler, D., M. J. Daly and E. S. Lander (2008). "Genetic mapping in human disease." Science 322(5903): 881-888. Alul, F. Y., D. E. Cook, O. A. Shchelochkov, L. G. Fleener, S. L. Berberich, J. C. Murray and K. K. Ryckman (2013a). "The heritability of metabolic profiles in newborn twins." Heredity 110(3): 253-258. Alul, F. Y., O. A. Shchelochkov, S. L. Berberich, J. C. Murray and K. K. Ryckman (2013b). "Genetic associations with neonatal thyroid-stimulating hormone levels." Pediatric research. Andersen, S., K. M. Pedersen, N. H. Bruun and P. Laurberg (2002). "Narrow individual variations in serum T(4) and T(3) in normal subjects: a clue to the understanding of subclinical thyroid disease." The Journal of clinical endocrinology and metabolism 87(3): 1068-1072. Appenzeller, S., A. Schirmacher, H. Halfter, S. Baumer, M. Pendziwiat, V. Timmerman, P. De Jonghe, et al. (2010). "Autosomal-dominant striatal degeneration is caused by a mutation in the phosphodiesterase 8B gene." American journal of human genetics 86(1): 83-87. Arnaud-Lopez, L., G. Usala, G. Ceresini, B. D. Mitchell, M. G. Pilia, M. G. Piras, N. Sestu, et al. (2008). "Phosphodiesterase 8B gene variants are associated with serum TSH levels and thyroid function." American journal of human genetics 82(6): 1270-1280. Barnett, I. J., S. Lee and X. Lin (2013). "Detecting rare variant effects using extreme phenotype sampling in sequencing association studies." Genetic epidemiology 37(2): 142-151. Berbel, P., D. Navarro, E. Auso, E. Varea, A. E. Rodriguez, J. J. Ballesta, M. Salinas, et al. (2010). "Role of late maternal thyroid hormones in cerebral cortex development: an experimental model for human prematurity." Cerebral cortex 20(6): 1462-1475. Boomsma, D., A. Busjahn and L. Peltonen (2002). "Classical twin studies and beyond." Nature reviews. Genetics 3(11): 872-882. Bosker, F. J., C. A. Hartman, I. M. Nolte, B. P. Prins, P. Terpstra, D. Posthuma, T. van Veen, et al. (2011). "Poor replication of candidate genes for major depressive disorder using genome-wide association data." Molecular psychiatry 16(5): 516-532. Cangul, H., Z. Aycan, A. Olivera-Nappa, H. Saglam, N. A. Schoenmakers, K. Boelaert, S. Cetinkaya, et al. (2012a). "Thyroid dyshormonogenesis is mainly caused by TPO mutations in consanguineous community." Clinical endocrinology. 111 Cangul, H., Z. Aycan, H. Saglam, J. R. Forman, S. Cetinkaya, O. Tarim, E. Bober, et al. (2012b). "TSHR is the main causative locus in autosomal recessively inherited thyroid dysgenesis." Journal of pediatric endocrinology & metabolism : JPEM 25(5-6): 419-426. Castanet, M. and M. Polak (2010). "Spectrum of Human Foxe1/TTF2 Mutations." Hormone research in paediatrics 73(6): 423-429. Center for Disease Control (2001). "Using tandem mass spectrometry for metabolic disease screening among newborns. A report of a work group." MMWR. Recommendations and reports : Morbidity and mortality weekly report. Recommendations and reports / Centers for Disease Control 50(RR-3): 1-34. Chace, D. H., J. C. DiPerna, B. L. Mitchell, B. Sgroi, L. F. Hofman and E. W. Naylor (2001). "Electrospray tandem mass spectrometry for analysis of acylcarnitines in dried postmortem blood specimens collected at autopsy from infants with unexplained cause of death." Clinical chemistry 47(7): 1166-1182. Chace, D. H., T. Lim, C. R. Hansen, V. R. De Jesus and W. H. Hannon (2009). "Improved MS/MS analysis of succinylacetone extracted from dried blood spots when combined with amino acids and acylcarnitine butyl esters." Clinica chimica acta; international journal of clinical chemistry 407(1-2): 6-9. Chiu, H. K., S. Sanda, P. Y. Fechner and C. Pihoker (2012). "Correlation of TSH with the risk of paediatric thyroid carcinoma." Clinical endocrinology 77(2): 316-322. Costa-e-Sousa, R. H. and A. N. Hollenberg (2012). "Minireview: The neural regulation of the hypothalamic-pituitary-thyroid axis." Endocrinology 153(9): 4128-4135. De Felice, M. and R. Di Lauro (2011). "Minireview: Intrinsic and extrinsic factors in thyroid gland development: an update." Endocrinology 152(8): 2948-2956. Denny, J. C., D. C. Crawford, M. D. Ritchie, S. J. Bielinski, M. A. Basford, Y. Bradford, H. S. Chai, et al. (2011). "Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenomewide studies." American journal of human genetics 89(4): 529-542. Dixon, M. J., M. L. Marazita, T. H. Beaty and J. C. Murray (2011). "Cleft lip and palate: understanding genetic and environmental influences." Nature reviews. Genetics 12(3): 167-178. Dube, J. B. and R. A. Hegele (2013). "Genetics 100 for cardiologists: basics of genomewide association studies." The Canadian journal of cardiology 29(1): 10-17. Dunham, I., A. Kundaje, S. F. Aldred, P. J. Collins, C. A. Davis, F. Doyle, C. B. Epstein, et al. (2012). "An integrated encyclopedia of DNA elements in the human genome." Nature 489(7414): 57-74. Emond, M. J., T. Louie, J. Emerson, W. Zhao, R. A. Mathias, M. R. Knowles, F. A. Wright, et al. (2012). "Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis." Nature genetics 44(8): 886-889. Epstein, D. J. (2009). "Cis-regulatory mutations in human disease." Briefings in functional genomics & proteomics 8(4): 310-316. 112 Fagman, H. and M. Nilsson (2010). "Morphogenesis of the thyroid gland." Molecular and cellular endocrinology 323(1): 35-54. Fagman, H. and M. Nilsson (2011). "Morphogenetics of early thyroid development." Journal of molecular endocrinology 46(1): R33-42. Ferrara, C. T., P. Wang, E. C. Neto, R. D. Stevens, J. R. Bain, B. R. Wenner, O. R. Ilkayeva, et al. (2008). "Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling." PLoS genetics 4(3): e1000034. Fleury, Y., G. Van Melle, V. Woringer, R. C. Gaillard and L. Portmann (2001). "Sexdependent variations and timing of thyroid growth during puberty." The Journal of clinical endocrinology and metabolism 86(2): 750-754. Gaitonde, D. Y., K. D. Rowley and L. B. Sweeney (2012). "Hypothyroidism: an update." American family physician 86(3): 244-251. Garduno-Garcia, J. J., U. Alvirde-Garcia, G. Lopez-Carrasco, M. E. Padilla Mendoza, R. Mehta, O. Arellano-Campos, R. Choza, et al. (2010). "TSH and free thyroxine concentrations are associated with differing metabolic markers in euthyroid subjects." European journal of endocrinology / European Federation of Endocrine Societies 163(2): 273-278. Gieger, C., L. Geistlinger, E. Altmaier, M. Hrabe de Angelis, F. Kronenberg, T. Meitinger, H. W. Mewes, et al. (2008). "Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum." PLoS genetics 4(11): e1000282. Goonewardena, S. N., L. E. Prevette and A. A. Desai (2010). "Metabolomics and atherosclerosis." Current atherosclerosis reports 12(4): 267-272. Grandone, A., L. Perrone, G. Cirillo, A. Di Sessa, A. M. Corona, A. Amato, N. Cresta, et al. (2012). "Impact of phosphodiesterase 8B gene rs4704397 variation on thyroid homeostasis in childhood obesity." European journal of endocrinology / European Federation of Endocrine Societies 166(2): 255-260. Grasberger, H. and S. Refetoff (2011). "Genetic causes of congenital hypothyroidism due to dyshormonogenesis." Current opinion in pediatrics 23(4): 421-428. Gudmundsson, J., P. Sulem, D. F. Gudbjartsson, J. G. Jonasson, G. Masson, H. He, A. Jonasdottir, et al. (2012). "Discovery of common variants associated with low TSH levels and thyroid cancer risk." Nature genetics 44(3): 319-322. Gudmundsson, J., P. Sulem, D. F. Gudbjartsson, J. G. Jonasson, A. Sigurdsson, J. T. Bergthorsson, H. He, et al. (2009). "Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations." Nature genetics 41(4): 460-464. Hannon, W. H., R. J. Whitley, B. Davin, P. Fernhoff, T. Halonen, M. Lavochkin, J. Miller, et al. (2007). "Blood Collection on Filter Paper for Newborn Screening Programs; Approved Standard-Fifth Edition." Clinical and Laboratory Standards Institute (document LA04-A5):Wayne, Pennsylvania, USA 27: 1-13. 113 Hansen, P. S., T. H. Brix, T. I. Sorensen, K. O. Kyvik and L. Hegedus (2004). "Major genetic influence on the regulation of the pituitary-thyroid axis: a study of healthy Danish twins." The Journal of clinical endocrinology and metabolism 89(3): 1181-1187. Hansen, P. S., W. M. van der Deure, R. P. Peeters, I. Iachine, M. Fenger, T. I. Sorensen, K. O. Kyvik, et al. (2007). "The impact of a TSH receptor gene polymorphism on thyroid-related phenotypes in a healthy Danish twin population." Clinical endocrinology 66(6): 827-832. Hayashi, M., K. Matsushima, H. Ohashi, H. Tsunoda, S. Murase, Y. Kawarada and T. Tanaka (1998). "Molecular cloning and characterization of human PDE8B, a novel thyroid-specific isozyme of 3',5'-cyclic nucleotide phosphodiesterase." Biochemical and biophysical research communications 250(3): 751-756. Hebrant, A., W. C. van Staveren, C. Maenhaut, J. E. Dumont and J. Leclere (2011). "Genetic hyperthyroidism: hyperthyroidism due to activating TSHR mutations." European journal of endocrinology / European Federation of Endocrine Societies 164(1): 1-9. Hinton, C. F., K. B. Harris, L. Borgfeld, M. Drummond-Borg, R. Eaton, F. Lorey, B. L. Therrell, et al. (2010). "Trends in incidence rates of congenital hypothyroidism related to select demographic factors: data from the United States, California, Massachusetts, New York, and Texas." Pediatrics 125 Suppl 2: S37-47. Hoermann, R. and J. E. Midgley (2012). "TSH Measurement and Its Implications for Personalised Clinical Decision-Making." Journal of thyroid research 2012: 438037. Horvath, A., V. Mericq and C. A. Stratakis (2008). "Mutation in PDE8B, a cyclic AMPspecific phosphodiesterase in adrenal hyperplasia." The New England journal of medicine 358(7): 750-752. Hwang, S. J., Q. Yang, J. B. Meigs, E. N. Pearce and C. S. Fox (2007). "A genome-wide association for kidney function and endocrine-related traits in the NHLBI's Framingham Heart Study." BMC medical genetics 8 Suppl 1: S10. Iliadou, A., P. Lichtenstein, U. de Faire and N. L. Pedersen (2001). "Variation in genetic and environmental influences in serum lipid and apolipoprotein levels across the lifespan in Swedish male and female twins." American journal of medical genetics 102(1): 48-58. Illig, T., C. Gieger, G. Zhai, W. Romisch-Margl, R. Wang-Sattler, C. Prehn, E. Altmaier, et al. (2010). "A genome-wide perspective of genetic variation in human metabolism." Nature genetics 42(2): 137-141. Jobling, R., R. A. Ferrier, R. McLeod, A. L. Petrin, J. C. Murray and M. A. Thomas (2011). "Monozygotic twins with variable expression of Van der Woude syndrome." American journal of medical genetics. Part A 155A(8): 2008-2010. Jones, A. M., K. M. Howarth, L. Martin, M. Gorman, R. Mihai, L. Moss, A. Auton, et al. (2012). "Thyroid cancer susceptibility polymorphisms: confirmation of loci on chromosomes 9q22 and 14q13, validation of a recessive 8q24 locus and failure to replicate a locus on 5q24." Journal of medical genetics 49(3): 158-163. 114 Julvez, J., M. Alvarez-Pedrerol, M. Rebagliato, M. Murcia, J. Forns, R. Garcia-Esteban, N. Lertxundi, et al. (2013). "Thyroxine levels during pregnancy in healthy women and early child neurodevelopment." Epidemiology 24(1): 150-157. Kaye, C. I., F. Accurso, S. La Franchi, P. A. Lane, N. Hope, P. Sonya, G. B. S, et al. (2006a). "Introduction to the newborn screening fact sheets." Pediatrics 118(3): 13041312. Kaye, C. I., F. Accurso, S. La Franchi, P. A. Lane, N. Hope, P. Sonya, G. B. S, et al. (2006b). "Newborn screening fact sheets." Pediatrics 118(3): e934-963. Kent, W. J., C. W. Sugnet, T. S. Furey, K. M. Roskin, T. H. Pringle, A. M. Zahler and D. Haussler (2002). "The human genome browser at UCSC." Genome research 12(6): 9961006. Kettunen, J., T. Tukiainen, A. P. Sarin, A. Ortega-Alonso, E. Tikkanen, L. P. Lyytikainen, A. J. Kangas, et al. (2012). "Genome-wide association study identifies multiple loci influencing human serum metabolite levels." Nature genetics 44(3): 269276. Keurentjes, J. J., J. Fu, C. H. de Vos, A. Lommen, R. D. Hall, R. J. Bino, L. H. van der Plas, et al. (2006). "The genetics of plant metabolism." Nature genetics 38(7): 842-849. Khoury, M. J., L. L. McCabe and E. R. McCabe (2003). "Population screening in the age of genomic medicine." The New England journal of medicine 348(1): 50-58. Kim, H. K., J. H. Yoon, S. J. Kim, J. S. Cho, S. S. Kweon and H. C. Kang (2013a). "Higher TSH level is a risk factor for differentiated thyroid cancer." Clinical endocrinology 78(3): 472-477. Kim, J. H., J. H. Park, S. Y. Kim and H. Y. Bae (2013b). "The mean platelet volume is positively correlated with serum thyrotropin concentrations in a population of healthy subjects and subjects with unsuspected subclinical hypothyroidism." Thyroid : official journal of the American Thyroid Association 23(1): 31-37. Kirsten, D. (2000). "The thyroid gland: physiology and pathophysiology." Neonatal network : NN 19(8): 11-26. Kratzsch, J. and F. Pulzer (2008). "Thyroid gland development and defects." Best practice & research. Clinical endocrinology & metabolism 22(1): 57-75. Kratzsch, J., G. Schubert, F. Pulzer, R. Pfaeffle, A. Koerner, A. Dietz, M. Rauh, et al. (2008). "Reference intervals for TSH and thyroid hormones are mainly affected by age, body mass index and number of blood leucocytes, but hardly by gender and thyroid autoantibodies during the first decades of life." Clinical biochemistry 41(13): 1091-1098. Kuhlenbaumer, G., P. Ludemann, A. Schirmacher, E. De Vriendt, G. Hunermund, P. Young, M. Hund-Georgiadis, et al. (2004). "Autosomal dominant striatal degeneration (ADSD): clinical description and mapping to 5q13-5q14." Neurology 62(12): 2203-2208. Kumar, J., R. Gordillo, F. J. Kaskel, C. M. Druschel and R. P. Woroniecki (2009). "Increased prevalence of renal and urinary tract anomalies in children with congenital hypothyroidism." The Journal of pediatrics 154(2): 263-266. 115 Kwon, C. and P. M. Farrell (2000). "The magnitude and challenge of false-positive newborn screening test results." Archives of pediatrics & adolescent medicine 154(7): 714-718. La Gamma, E. F. and N. Paneth (2012). "Clinical importance of hypothyroxinemia in the preterm infant and a discussion of treatment concerns." Current opinion in pediatrics 24(2): 172-180. LaFranchi, S. (1999). "Thyroid function in the preterm infant." Thyroid : official journal of the American Thyroid Association 9(1): 71-78. Lakics, V., E. H. Karran and F. G. Boess (2010). "Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues." Neuropharmacology 59(6): 367-374. Lamina, C. (2011). "Digging into the extremes: a useful approach for the analysis of rare variants with continuous traits?" BMC proceedings 5 Suppl 9: S105. Leger, J., D. Marinovic, C. Garel, C. Bonaiti-Pellie, M. Polak and P. Czernichow (2002). "Thyroid developmental anomalies in first degree relatives of children with congenital hypothyroidism." The Journal of clinical endocrinology and metabolism 87(2): 575-580. Leonardi, D., N. Polizzotti, A. Carta, R. Gelsomino, L. Sava, R. Vigneri and F. Calaciura (2008). "Longitudinal study of thyroid function in children with mild hyperthyrotropinemia at neonatal screening for congenital hypothyroidism." The Journal of clinical endocrinology and metabolism 93(7): 2679-2685. Lowe, J. K., J. B. Maller, I. Pe'er, B. M. Neale, J. Salit, E. E. Kenny, J. L. Shea, et al. (2009). "Genome-wide association studies in an isolated founder population from the Pacific Island of Kosrae." PLoS genetics 5(2): e1000365. Manolio, T. A. (2010). "Genomewide association studies and assessment of the risk of disease." The New England journal of medicine 363(2): 166-176. Manolio, T. A., F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff, D. J. Hunter, M. I. McCarthy, et al. (2009). "Finding the missing heritability of complex diseases." Nature 461(7265): 747-753. Medici, M., W. M. van der Deure, M. Verbiest, S. H. Vermeulen, P. S. Hansen, L. A. Kiemeney, A. R. Hermus, et al. (2011). "A large-scale association analysis of 68 thyroid hormone pathway genes with serum TSH and FT4 levels." European journal of endocrinology / European Federation of Endocrine Societies 164(5): 781-788. Michel, S., L. Liang, M. Depner, N. Klopp, A. Ruether, A. Kumar, M. Schedel, et al. (2010). "Unifying candidate gene and GWAS Approaches in Asthma." PloS one 5(11): e13894. Midgley, J. E., R. Hoermann, R. Larisch and J. W. Dietrich (2013). "Physiological states and functional relation between thyrotropin and free thyroxine in thyroid health and disease: in vivo and in silico data suggest a hierarchical model." J Clin Pathol 19: 19. Mihalik, S. J., B. H. Goodpaster, D. E. Kelley, D. H. Chace, J. Vockley, F. G. Toledo and J. P. DeLany (2010). "Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity." Obesity 18(9): 1695-1700. 116 National Newborn Screening and Genetics Resource Center. (2013). February, 2013, from http://genes-r-us.uthscsa.edu/. Retrieved Nicholson, G., M. Rantalainen, A. D. Maher, J. V. Li, D. Malmodin, K. R. Ahmadi, J. H. Faber, et al. (2011). "Human metabolic profiles are stably controlled by genetic and environmental variation." Molecular systems biology 7: 525. Nyholt, D. R. (2006). "On the probability of dizygotic twins being concordant for two alleles at multiple polymorphic loci." Twin research and human genetics : the official journal of the International Society for Twin Studies 9(2): 194-197. Online Mendelian Inheritance in Man. (2013). http://omim.org/. Retrieved February, 2013, from Panicker, V. (2011). "Genetics of thyroid function and disease." The Clinical biochemist. Reviews / Australian Association of Clinical Biochemists 32(4): 165-175. Panicker, V., S. G. Wilson, T. D. Spector, S. J. Brown, M. Falchi, J. B. Richards, G. L. Surdulescu, et al. (2008). "Heritability of serum TSH, free T4 and free T3 concentrations: a study of a large UK twin cohort." Clinical endocrinology 68(4): 652-659. Panicker, V., S. G. Wilson, J. P. Walsh, J. B. Richards, S. J. Brown, J. P. Beilby, A. P. Bremner, et al. (2010). "A locus on chromosome 1p36 is associated with thyrotropin and thyroid function as identified by genome-wide association study." American journal of human genetics 87(3): 430-435. Parlato, R., A. Rosica, A. Rodriguez-Mallon, A. Affuso, M. P. Postiglione, C. Arra, A. Mansouri, et al. (2004). "An integrated regulatory network controlling survival and migration in thyroid organogenesis." Developmental biology 276(2): 464-475. Pass, K. A. and E. C. Neto (2009). "Update: newborn screening for endocrinopathies." Endocrinology and metabolism clinics of North America 38(4): 827-837. Porcu, E., M. Medici, G. Pistis, C. B. Volpato, S. G. Wilson, A. R. Cappola, S. D. Bos, et al. (2013). "A meta-analysis of thyroid-related traits reveals novel Loci and genderspecific differences in the regulation of thyroid function." PLoS genetics 9(2): e1003266. Purcell, S., B. Neale, K. Todd-Brown, L. Thomas, M. A. Ferreira, D. Bender, J. Maller, et al. (2007). "PLINK: a tool set for whole-genome association and population-based linkage analyses." American journal of human genetics 81(3): 559-575. Rastogi, M. V. and S. H. LaFranchi (2010). "Congenital hypothyroidism." Orphanet journal of rare diseases 5: 17. Rawal, R., A. Teumer, H. Volzke, H. Wallaschofski, T. Ittermann, B. O. Asvold, T. Bjoro, et al. (2012). "Meta-analysis of two genome-wide association studies identifies four genetic loci associated with thyroid function." Human molecular genetics 21(14): 3275-3282. Roef, G., B. Lapauw, S. Goemaere, H. Zmierczak, T. Fiers, J. M. Kaufman and Y. Taes (2011). "Thyroid hormone status within the physiological range affects bone mass and density in healthy men at the age of peak bone mass." European journal of endocrinology / European Federation of Endocrine Societies 164(6): 1027-1034. 117 Roos, A., S. J. Bakker, T. P. Links, R. O. Gans and B. H. Wolffenbuttel (2007). "Thyroid function is associated with components of the metabolic syndrome in euthyroid subjects." The Journal of clinical endocrinology and metabolism 92(2): 491-496. Rose, S. R., R. S. Brown, T. Foley, P. B. Kaplowitz, C. I. Kaye, S. Sundararajan and S. K. Varma (2006). "Update of newborn screening and therapy for congenital hypothyroidism." Pediatrics 117(6): 2290-2303. Rosenbloom, K. R., C. A. Sloan, V. S. Malladi, T. R. Dreszer, K. Learned, V. M. Kirkup, M. C. Wong, et al. (2013). "ENCODE data in the UCSC Genome Browser: year 5 update." Nucleic acids research 41(Database issue): D56-63. Samollow, P. B., G. Perez, C. M. Kammerer, D. Finegold, P. W. Zwartjes, L. M. Havill, A. G. Comuzzie, et al. (2004). "Genetic and environmental influences on thyroid hormone variation in Mexican Americans." The Journal of clinical endocrinology and metabolism 89(7): 3276-3284. Sawin, C. T., A. Geller, P. A. Wolf, A. J. Belanger, E. Baker, P. Bacharach, P. W. Wilson, et al. (1994). "Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons." The New England journal of medicine 331(19): 1249-1252. Schmaltz, C. (2012). "Thyroid hormones in the neonate: an overview of physiology and clinical correlation." Advances in neonatal care : official journal of the National Association of Neonatal Nurses 12(4): 217-222. Shah, A. A., D. M. Craig, J. K. Sebek, C. Haynes, R. C. Stevens, M. J. Muehlbauer, C. B. Granger, et al. (2012). "Metabolic profiles predict adverse events after coronary artery bypass grafting." The Journal of thoracic and cardiovascular surgery 143(4): 873-878. Shah, S. H., J. R. Bain, M. J. Muehlbauer, R. D. Stevens, D. R. Crosslin, C. Haynes, J. Dungan, et al. (2010). "Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events." Circulation. Cardiovascular genetics 3(2): 207-214. Shah, S. H., E. R. Hauser, J. R. Bain, M. J. Muehlbauer, C. Haynes, R. D. Stevens, B. R. Wenner, et al. (2009). "High heritability of metabolomic profiles in families burdened with premature cardiovascular disease." Molecular systems biology 5: 258. Shields, B. M., R. M. Freathy, B. A. Knight, A. Hill, M. N. Weedon, T. M. Frayling, A. T. Hattersley, et al. (2009). "Phosphodiesterase 8B gene polymorphism is associated with subclinical hypothyroidism in pregnancy." The Journal of clinical endocrinology and metabolism 94(11): 4608-4612. Shields, B. M., B. A. Knight, A. Hill, A. T. Hattersley and B. Vaidya (2011). "Fetal thyroid hormone level at birth is associated with fetal growth." The Journal of clinical endocrinology and metabolism 96(6): E934-938. Srinivasan, M. and M. S. Patel (2008). "Metabolic programming in the immediate postnatal period." Trends in endocrinology and metabolism: TEM 19(4): 146-152. Stathatos, N. (2012). "Thyroid physiology." The Medical clinics of North America 96(2): 165-173. 118 Steffen, K. M., M. E. Cooper, M. Shi, D. Caprau, H. N. Simhan, J. M. Dagle, M. L. Marazita, et al. (2007). "Maternal and fetal variation in genes of cholesterol metabolism is associated with preterm delivery." Journal of perinatology : official journal of the California Perinatal Association 27(11): 672-680. Suhre, K., S. Y. Shin, A. K. Petersen, R. P. Mohney, D. Meredith, B. Wagele, E. Altmaier, et al. (2011a). "Human metabolic individuality in biomedical and pharmaceutical research." Nature 477(7362): 54-60. Suhre, K., H. Wallaschofski, J. Raffler, N. Friedrich, R. Haring, K. Michael, C. Wasner, et al. (2011b). "A genome-wide association study of metabolic traits in human urine." Nature genetics 43(6): 565-569. Szkudlinski, M. W., V. Fremont, C. Ronin and B. D. Weintraub (2002). "Thyroidstimulating hormone and thyroid-stimulating hormone receptor structure-function relationships." Physiological reviews 82(2): 473-502. Targovnik, H. M., S. A. Esperante and C. M. Rivolta (2010). "Genetics and phenomics of hypothyroidism and goiter due to thyroglobulin mutations." Molecular and cellular endocrinology 322(1-2): 44-55. Teumer, A., R. Rawal, G. Homuth, F. Ernst, M. Heier, M. Evert, F. Dombrowski, et al. (2011). "Genome-wide association study identifies four genetic loci associated with thyroid volume and goiter risk." American journal of human genetics 88(5): 664-673. Tomaz, R. A., I. Sousa, J. G. Silva, C. Santos, M. R. Teixeira, V. Leite and B. M. Cavaco (2012). "FOXE1 polymorphisms are associated with familial and sporadic nonmedullary thyroid cancer susceptibility." Clinical endocrinology 77(6): 926-933. Turgeon, C., M. J. Magera, P. Allard, S. Tortorelli, D. Gavrilov, D. Oglesbee, K. Raymond, et al. (2008). "Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots." Clinical chemistry 54(4): 657-664. UCSC Genome Browser. Retrieved March, 2013, from http://genome.ucsc.edu/. van der Deure, W. M., A. G. Uitterlinden, A. Hofman, F. Rivadeneira, H. A. Pols, R. P. Peeters and T. J. Visser (2008). "Effects of serum TSH and FT4 levels and the TSHRAsp727Glu polymorphism on bone: the Rotterdam Study." Clinical endocrinology 68(2): 175-181. Vanderpump, M. P. (2011). "The epidemiology of thyroid disease." British medical bulletin 99: 39-51. Votava, F., D. Torok, J. Kovacs, D. Moslinger, S. M. Baumgartner-Parzer, J. Solyom, Z. Pribilincova, et al. (2005). "Estimation of the false-negative rate in newborn screening for congenital adrenal hyperplasia." European journal of endocrinology / European Federation of Endocrine Societies 152(6): 869-874. Vulsma, T., M. H. Gons and J. J. de Vijlder (1989). "Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis." The New England journal of medicine 321(1): 13-16. 119 Wang, T. J., M. G. Larson, R. S. Vasan, S. Cheng, E. P. Rhee, E. McCabe, G. D. Lewis, et al. (2011). "Metabolite profiles and the risk of developing diabetes." Nature medicine 17(4): 448-453. Watson, M. S., M. Y. Mann, M. A. Lloyd-Puryear, P. Rinaldo and R. R. Howell (2006). "Newborn screening: toward a uniform screening panel and system—executive summary." Pediatrics 117(Supplement 3): S296-S307. Wilcken, B. and V. Wiley (2008). "Newborn screening." Pathology 40(2): 104-115.