Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Decoding ENCODE
Jim Kent
University of California Santa Cruz
ENCODE Timeline
• ENCyclopedia of Dna Elements.
– Attempt to catalog as many functional
elements in human genome as possible
using current technologies.
– Pilot project - finished 2007, covered 1% of
genome.
– Production project - ramping up now.
Genome-wide. Should have major
amounts of data in 6 months.
ENCODE Experiments
• Chromatin state:
– DNA Hypersensitivity assays
– Chromatin Immunoprecipitation (ChIP)
• Histones in various methylation states
• Sequence-specific transcription factors
– DNA methylation
– Chromatin conformation capture (5C)
• Functional RNA discovery
– Nuclear & cytoplasmic, short & long
– RNA Immunoprecipitation
• Comparative Genomics
• Human curated gene annotation
Role of UCSC
• Display data in context of what else is
known on the UCSC Genome Browser
and in other tools.
• Facilitate analysis of the data with both
Web-based and command line tools.
A Peek at the Pilot Project
ENCODE pilot data at genome.ucsc.edu
Correlation at gene starts in
enr221
Transcription at enm221
ENCODE Chromatin
Immunoprecipitation
Scientific Highlights of Pilot
• Transcription:
– Lots of transcription outside of known genes.
– Outside of known genes transcribed areas not
very well conserved across species.
– Lots of rare splice variants, also poorly conserved.
• DNA/Protein Interactions
– Good correlation between histone markers, gene
starts, and _active_ transcription.
– Lots of “occupied transcription factor binding sites”
not conserved, near promoters etc.
• Biological noise?
– Main controversy was whether to explain much of
the data as “biological noise” that was tolerated
but not necessary for function.
From Pilot to Production Phase
ENCODE Production Phase
• Moving from microarray based assays
to assays based on next-generation
sequencing. (ChIP-chip to ChIP-seq)
• Genome-wide rather than regional.
• Broader set of cell lines used more
consistently between labs.
• Broader set of antibodies.
• Some new technology development
continues.
ENCODE Cell Lines
• Tier 1 - used in ALL experiments
– GM12878 (lymphoblastoid cell line)
– K562 (chronic myeloid leukemia)
• Tier 2 - used in most experiments
–
–
–
–
–
HepG2 (hepatocellular carcinoma)
Hela-S3 (cervical carcinoma)
HUVEC (umbilical vein endothelial cells)
Keratinocyte (normal epidermal cells)
Likely will do an embryonic stem cell too.
• Tier 3 - used in one or two experiments
– Many of these for assays such as DNAse
hypersensitivity, RNA measurements where don’t
have to do separate experiment for each antibody.
Simple Model of Eukaryotic
Transcription Regulation
• Initially chromatin “opened” to allow
transcription factors to access DNA
• Multiple transcription factors bind to DNA in
combination.
– Most factors have such small DNA binding sites
that by themselves they are not specific or the
binding even stable
• The right combination of factors in open chromatin
leads to active transcription starting at the initiation
complex.
• With the ENCODE experiments we can directly test
most aspects of this model.
Chromatin Experiments
• In general applied across a large number of
cell lines.
• DNAseI hypersensitivity
• Formaldehyde Assisted Isolation of
Regulatory Elements
• Methylation of CpG Islands
• ChIP-seq of relevant factors
– H3K4me1,2,3 H3K9me3 H4K20me3, H3K27me3,
H3K36me3, RPol-II, etc.
Transcription Factor ChIP
• Many antibodies in modest number of cell
lines.
• Limited by good antibodies, hope for 100 or
more.
• Current good antibodies include
– E2F1, E2F4, E2F6, KAP1, L3MBTL2, STAT1,
CtBP1, CtBP2, SETDB1, ZNF180, ZNF239,
ZNF263, ZNF266, ZNF317, ZNF342
• Part of project pipeline for raising and testing
antibodies.
RNA measurement
• RNA-seq of poly-A selected RNA to measure
mRNA levels in many cell lines.
• Sequencing of G-cap selected tags (CAGE)
• Sequencing 5’ and 3’ ends (paired end tags)
• Measurement of RNAs of several types in
several cell compartments of a few cell lines.
– Long/short, polyA/nonPolyA, associated with
proteins/not associated with proteins
– Nucleus, cytosol, polysomes, chromatin, nucleolus
New Pilot Projects
Starting to Sprout
New Pilot Projects
• Immunoprecipitation of RNA binding
proteins/RNA sequencing.
• Mapping silencers and enhancers with
transient transfection assays
• Computational identification of active
promoters
• Deep comparative sequencing in targeted
regions and conservation analysis.
• Chromatin Conformation Capture Carbon
Copy (5C) to capture long range regulatory
elements and their targets.
ENCODE Timeline
• Grants funded for 4 years starting Sept
2007.
• First production data just now starting to
roll into UCSC, not quite ready for public
display.
• Data should accumulate quickly over
next few years.
Data Release Policy
• Once have reproducible data (where at least
2 of 3 replicates agree) should be released to
public within a month.
• Data is still considered pre-publication!
– Ok to publish a paper using data on a few genes.
– Please wait for consortium papers before papers
doing full genome analysis.
– Anyone can join ENCODE consortium analysis
group to help us write the papers.
– We just have ~1 year after data release to write
papers, after that fair game to publish full genome
analysis.
– If in doubt please contact consortium via UCSC.
Web Works for Mice and Men
Mouse ES Cell Chromatin IP
• Brad Bernstein lab ChIP-seq based
experiment on methylated histones now
on UCSC Genome Browser.
• Shows some of the user interfaces that
will be used for the ENCODE data
List of mouse chromatin subtracks….
Signal densities of entire mouse chromatin data
set.
The unending quest for genes
Gencode Project
• Project to define structure (exons and introns)
for all common splice varients of all genes.
• Human curators merge many lines of
evidence including
–
–
–
–
–
Computational gene predictions
RNA/DNA alignments
Paired end tags
Cross-species alignments
Possibly chromatin state data
• PI is Tim Hubbard
• Much of the work done by Havana group
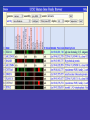
Data Mining with Table
Browser
QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture.
Table Browser
• Complete access to UCSC Database
with results in tab-delimited format
• Method for creating “custom tracks” by
combining and filtering existing tracks.
• Sample query - getting a table of
Ensembl gene coordinates and
associated Superfamily annotations.
Selected fields from related tables results: Ensemble Gene
(ensGene) and Superfamily Description (sfDescription).
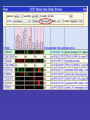
Table Browser Filters
• Getting list of Ensembl genes that have
SH3 domains.
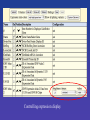
Table Browser Intersection
• Getting list of Ensembl genes that don’t
intersect UCSC Known Genes
Custom Track Output
• Useful for visualizing results of queries
in genome browser
• The way to produce more complex
queries.
• Here we look at how well genes that are
Ensembl but not UCSC are conserved
across species.
681/3329 (20%) of Ensemble not known also not conserved
1728/33,666 (5%) of Ensembl in general not conserved
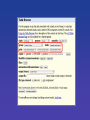
UCSC Gene Sorter
Quick Time™a nd a TIFF ( Uncomp res sed) deco mpre ssor are n eede d to s ee this picture .
• Swiss army knife for dealing with gene
sets.
• Hilights relationships and connections
between genes.
• Powerful data mining tool.
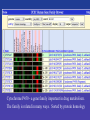
Cytochrome P450 - a gene family important in drug metabolism.
The family is related in many ways. Sorted by protein homology
Various sorting methods let you focus on different types
of relationships between genes.
Sorting by gene distance is a quick way to browse candidate
genes in a region.
Clicking on row # or gene name selects that gene.
Configuration page controls column order and display options.
Also you can upload your own columns here.
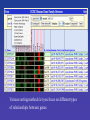
Controlling expression display
GNF Atlas 2 column in ‘median of replicates’ mode. Actual
Column includes 79 tissues, slide only fits first half.
Sorting based on expression similarity to selected gene.
The filters page turns the Family Browser into a powerful
data mining tool.
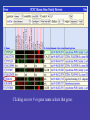
Candidate Pancreatic Islet Membrane Genes
GO-annotated membrane proteins that are expressed at
least 8X in pancreatic islets cells and no more than 4X
elsewhere outside of pancreas and central nervous system.
These might be good candidates for targets of the
autoimmune response that can cause Type I diabetes.
Direct Data Access
FTP or HTTP Download
•
•
•
•
•
Sequence
Multiple genome alignments
“Wiggle” track data.
Database as tab-separated files
Follow downloads link from
http://genome.ucsc.edu
• Via ftp://hgdownload.cse.ucsc.edu
Public MySQL Access
• Query mirror of our database directly
– Host: genome-mysql.cse.ucsc.edu
– User: genome
– No password needed
• Best to use table browser to find relevant
tables in many cases.
• Some tables are split by chromosomes
– chr1_est, chr2_est, etc.
• Some data (genome sequence, multiple
alignments, wiggles) are in files just
referenced by SQL tables.
• For some purposes easier to use via UCSC C
library code than via SQL.
The Sordid Details of the
UCSC Genome Informatics
Code Base
Download via http://genome.ucsc.edu/admin/cvs.html
Many modules require MySQL to be installed.
Lagging Edge Software
• C language - compilers still available!
• CGI Scripts - portable if not pretty.
• SQL database - at least MySQL is free.
Problems with C
• Missing booleans and
strings.
• No real objects.
• Must free things
Coping with Missing Data
Types in C
• #define boolean int
• Fixing lack of real string type much
harder
– lineFile/common modules and autoSql
code generator make parsing files
relatively painless
– dyString module not a horrible string ‘class’
Object Oriented Programming
in C
• Build objects around structures.
• Make families of functions with names that
start with the structure name, and that take
the structure as the first argument.
• Implement polymorphism/virtual functions
with function pointers in structure.
• Inheritance is still difficult. Perhaps this is not
such a bad thing.
struct dnaSeq
/* A dna sequence in one-letter-per-base format. */
{
struct dnaSeq *next; /* Next in list. */
char *name;
/* Sequence name. */
char *dna;
/* a’s c’s g’s and t’s. Null terminated */
int size;
/* Number of bases. */
};
struct dnaSeq *dnaSeqFromString(char *string);
/* Convert string containing sequence and possibly
* white space and numbers to a dnaSeq. */
void dnaSeqFree(struct dnaSeq **pSeq);
/* Free dnaSeq and set pointer to NULL. */
void dnaSeqFreeList(struct dnaSeq **pList);
/* Free list of dnaSeq’s. */
struct screenObj
/* A two dimensional object in a sleazy video game. */
{
struct screenObj *next; /* Next in list. */
char *name;
/* Object name. */
int x,y,width,height; /* Bounds of object. */
void (*draw)(struct screenObj *obj); /* Draw object */
boolean (*in)(struct screenObj *obj, int x, int y);
/* Return true if x,y is in object */
void *custom; /* Custom data for a particular type */
void (*freeCustom)(struct screenObj *obj);
/* Free custom data. */
};
#define screenObjDraw(obj) (obj->draw(obj))
/* Draw object. */
void screenObjFree(struct screenObj **pObj);
/* Free up screen object including custom part. */
Relational Databases
• Relational databases consist of tables, indices, and
the Structured Query Language (SQL).
• Tables are much like tab-separated files:
#chrom
chr22
chr21
start
end
14600000 14612345
18283999 18298577
name
ldlr
vldlr
strand
+
-
score
0.989
0.998
Fields are simple - no lists or substructures.
• Can join tables based on a shared field. This is
flexible, but only as fast as the index.
• Tables and joins are accessed a row at a time.
• The row is represented as an array of strings.
Converting A Row to Object
struct exoFish *exoFishLoad(char **row)
/* Load a exoFish from row fetched with select * from exoFish
* from database. Dispose of this with exoFishFree(). */
{
struct exoFish *ret;
AllocVar(ret);
ret->chrom = cloneString(row[0]);
ret->chromStart = sqlUnsigned(row[1]);
ret->chromEnd = sqlUnsigned(row[2]);
ret->name = cloneString(row[3]);
ret->score = sqlUnsigned(row[4]);
return ret;
}
Motivation for AutoSql
• Row to object code is tedious at best.
• Also have save object, free object code
to write.
• SQL create statement needs to match C
structure.
• Lack of lists without doing a join can
seriously impact performance and
complicate schema.
AutoSql Data Declaration
table exoFish
"An evolutionarily conserved region (ecore) with Tetroadon"
(
string chrom;
"Human chromosome or FPC contig"
uint chromStart; "Start position in chromosome"
uint chromEnd; "End position in chromosome"
string name;
"Ecore name in Genoscope database"
uint score;
"Score from 0 to 1000"
)
See autoSql.doc for more details.
Occasionally useful tools
Unix Command Line
• BLAT - RNA/DNA and DNA/DNA alignment.
• featureBits - figure out number of bases
covered by a track or intersection of tracks,
output track intersections.
• htmlCheck - check html tables and other
basic web page stuff. Look at form variables.
• dbSnoop - summarize a MySQL database.
• autoSql - generate serialization C code for
relational databases/tab-separated files.
• autoXml - generate XML parsers
• xmlToSql/sqlToXml - convert between XML
and relational database representations
• parasol - manage jobs on computer cluster
C Library Modules
• hdb - access UCSC genome database
• jksql - access SQL databases
• htmlPage - parse web pages, submit
forms
• readers/writers for maf, psl, chain, net,
bed, 2bit other formats used at UCSC
• rangeTree & binRange - fast interval
intersection tools
• Hashes, lists, trees, etc.