Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
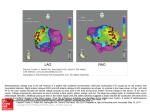
Cardiovascular Research 46 (2000) 250–256 www.elsevier.com / locate / cardiores www.elsevier.nl / locate / cardiores Review Infarct scar: a dynamic tissue Yao Sun, Karl T. Weber* Division of Cardiovascular Diseases, Department of Internal Medicine, University of Tennessee, Memphis College of Medicine, Rm. 353 Dobbs Research Institute, 951 Court Avenue, Memphis, TN 38163, USA Received 23 November 1999; accepted 21 January 2000 Abstract Infarct scar, a requisite to the rebuilding of necrotic myocardium following myocardial infarction (MI), has long been considered inert. Earlier morphologic studies suggested healing at the infarct site was complete within 6–8 weeks following MI and resultant scar tissue, albeit necessary, was acellular and simply fibrillar collagen. Utilizing molecular and cellular biologic technologies, recent studies indicate otherwise. Infarct scar is composed of phenotypically transformed fibroblast-like cells, termed myofibroblasts (myoFb) because they express alpha-smooth muscle actin (a-SMA) and these microfilaments confer contractile behavior in response to various peptides and amines. These cells are nourished by a neovasculature and are persistent at the MI site, where they are metabolically active expressing components requisite to angiotensin (Ang) peptide generation, including converting enzyme, receptors for AngII and transforming growth factor (TGF)-b1. They continue to elaborate fibrillar type I collagen. Their generation of these peptides contribute to ongoing scar tissue collagen turnover and to fibrous tissue formation of noninfarcted myocardium. Infarct scar contraction accounts for its thinning and its tonus may contribute to abnormal ventricular chamber stiffness with diastolic dysfunction. Infarct scar is a dynamic tissue: cellular, vascularized, metabolically active and contractile. Pharmacologic interventions with angiotensin converting enzyme inhibitor or AT1 receptor antagonist has proven effective in attenuating scar tissue metabolic activity and minimizing adverse accumulation of fibrous tissue in noninfarcted myocardium. 2000 Elsevier Science B.V. All rights reserved. Keywords: Angiotensin; Fibrosis; Growth factors; Infarction 1. Introduction Following MI with loss of necrotic cardiac myocytes, a reparative process is quickly initiated to rebuild infarcted myocardium and maintain structural integrity of the ventricle. A series of cellular responses are called into play driven largely by cell–cell signaling that serves to regulate tissue repair. Initially, inflammatory cells are attracted to and invade the site of injury, regulatory peptides are activated and elaborated, new blood vessels are formed (angiogenesis), and fibroblast-like cells appear and replicate. This early inflammatory phase of healing with resultant granulation tissue formation is followed by a fibrogenic phase that eventuates in scar tissue — a rebuilding of infarcted myocardium. In the case of a large transmural MI, the entire heart is involved in the repair *Corresponding author. Tel.: 11-901-448-5759; fax: 11-901-4488084. E-mail address: [email protected] (K.T. Weber) process with unwanted fibrous tissue appearing at sites remote to the MI and contributing to a remodeling of noninfarcted myocardium. Postinfarction healing has been considered complete 6–8 weeks following MI. Moreover, the infarct scar is viewed as inert tissue — simply fibrillar, cross-linked collagen whose tensile strength resists deformation and rupture. Accordingly, there has been little interest in scar tissue and any active role it may play in the failing heart of ischemic origins. Jugdutt et al. [1,2] have systematically examined the topography and temporal response in the architectural remodeling of infarct scar following MI of the canine heart. Scar thinning was observed at 6–8 weeks [1]. This would suggest an active process of scar tissue contraction had occurred. In recent years and using technologies of molecular and cellular biology, a new perspective of the infarct scar has emerged. One that reveals a cellularity based on a populaTime for primary review 27 days. 0008-6363 / 00 / $ – see front matter 2000 Elsevier Science B.V. All rights reserved. PII: S0008-6363( 00 )00032-8 Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 tion of fibroblast-like cells, termed myofibroblasts (myoFb), nourished by a neovasculature and whose metabolic activity includes generating peptides that regulate ongoing type I collagen synthesis in an autocrine manner and whose a-SMA microfilaments and attachments to extracellular matrix confers contractile behavior to scar tissue [3–5]. This review focuses on these features of infarct scar tissue and its dynamic nature. 2. Rebuilding and remodeling myocardium following infarction 2.1. Collagen turnover in infarct scar Cardiac tissue consists of a muscular compartment composed of large cardiac myocytes and an interstitial compartment that contains extracellular matrix and vasculature, each having their own distinctive cellular composition. Transmural MI involves a segmental loss of cardiac myocytes. Tissue repair must follow to rebuild and restore structural integrity at the infarct site. The balance between collagen synthesis and degradation are primary determinants of tissue fibrosis. Collagen degradation involves proteolytic enzymes (or matrix metalloproteinases, MMPs). MMP-1 (or collagenase) and MMP-8 (a gelatinase) degrade fibrillar collagen into fragments, which are further degraded into amino acids and oligopeptides by MMP-2, 3, and 9 [6]. During the very early phase of repair that follows MI in rats, degradation predominates represented as an initial increase in MMP-1 activity and its subsequent mRNA expression [7,8]. This early proteolytic activity accounts for fibrillar collagen degradation at the site of MI. Tissue inhibitors of MMPs (TIMPs) neutralize this collagenolytic activity and function as a regulatory brake on the activity of MMPs. TIMPs directly inhibit the activated form of MMPs. TIMP synthesis at the infarct site is elevated during week 1 and in subsequent weeks [7], which suppresses the activity of MMPs in the infarcted rat myocardium and promotes progressive collagen accumulation. Scar tissue is composed predominantly of type I and III fibrillar collagens. Temporal response of cardiac collagen turnover has been examined in rats with MI created by left coronary artery ligation. By northern blot and in situ hybridization analyses, type I procollagen mRNA at infarction site increases soon after MI and remains elevated over the course of 3 months (Fig. 1) [9]. This is an extended period of time based on the normal 2–3 year life-span of rats. Microscopic evidence of collagen fiber accumulation appears at the border zone to the infarct as early as day 7. An organized assembly of fibers in the form of scar tissue becomes evident by day 14 and continues to accumulate for many weeks [10,11]. Hydroxyproline concentration at the site of scarring increases progressively for over 6 weeks [1]. These findings lend support to the concept that 251 collagen is continuously synthesized and deposited in the infarct scar. Unlike traditional concepts, fibrous tissue formation in the infarcted heart is not a transient process but rather an ongoing one. 2.2. Collagen turnover at remote sites Following large anterior transmural MI in rats, fibrous tissue also appears in noninfarcted myocardium, but to a lesser extent than seen at the site of MI [12–15]. Expression of procollagen I and III mRNAs by fibroblast-like cells is increased in the noninfarcted interventricular septum and right ventricle on days 4 and 7, respectively. In the septum closest to the anterior MI, type I collagen mRNA remains elevated until day 28. In the right ventricle, more distant to the infarct site, message for these collagens is attenuated after day 7. Interstitial collagen appears at each of these remote sites to create a remodeling of noninfarcted myocardium by day 14 and continues to accumulate for weeks. Fibroblast-like cells are involved in collagen turnover at these sites. Right ventricle stiffness is significantly increased 8 weeks following anterior MI in rats. 3. Myofibroblasts and infarct scar The growth and activity of extracellular matrix producing cells are integral to tissue fibrosis. Interstitial fibroblasts are responsible for collagen synthesis in the normal myocardium, while phenotypically transformed fibroblasts, termed myoFb, are central to fibrogenesis at sites of rebuilding and remodeling following MI [16,17]. MyoFb are not residents of normal cardiac tissue (except heart valve leaflets). They appear at the infarct site. A hallmark of myoFb is their expression of a-SMA microfilaments. MyoFb appear to arise from interstitial fibroblasts and / or adventitial fibroblasts [18,19], however, progenitor cells are presently uncertain. Signals that determine the appearance of myoFb are not fully understood. In vivo and in vitro studies reveal that transforming growth factor-beta (TGF-b1) contributes to fibroblast differentiation into myoFb [20]. Activated macrophages appear at the site of MI on days 1 and 2, where they elaborate TGF-b1. MyoFb appear at the site of infarction thereafter. In experimental MI in rats, myoFb first appear at the site of MI as early as day 3, become evident at week 1 and remain abundant for months thereafter. These cells are colocalized with accumulated collagen. They persist in the infarct scar for prolonged periods of time (many months in rats, years in man) (Fig. 1) [16,21], where they continue to generate fibrogenic signals that perpetuate tissue repair and promote fibrosis. By in situ hybridization, myoFb are responsible for increased expression of genes encoding for fibrillar type I / III procollagens [9,22]. Why myoFb persist in the infarct heart has not been elucidated. This contrasts to skin, 252 Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 Fig. 1. Eight weeks following MI, the infarct scar still contains abundant myofibroblasts (MF) (panel A, a-SMA labeling) and remains vascularized (panel B, a-SMA labeling at vessels labeled with arrowheads); type I collagen and TGF-b1 mRNA (panels, C and D, in situ hybridization) expressions are still elevated at the site of MI; and binding density of ACE and AngII receptors (panels E and F, autoradiography) continues to be increased at this site. RV, right ventricle. where they disappear (via apoptosis) once healing is complete [23]. Their persistence in other injured organs is associated with a progressive fibrosis and predicts organ failure. In an experimental model of toxic nephritis, progressive renal fibrosis is seen only in those kidneys in which myoFb fail to disappear [24]. In humans with Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 immunoglobulin A nephropathy, the evidence of a-SMA labeling on renal biopsy tissue is a predictor of poor renal function and correlates with progressive renal fibrosis [25]. The importance of persistent fibrogenic signals that perpetuate tissue repair and an unwanted fibrosis is emphasized not only within a particular organ, but could take on even broader implications for multiple tissues when such signals are chronically elevated and not neutralized. 4. Neovasculature and infarct scar Angiogenesis is a major feature of tissue repair. Following MI, angiogenesis begins in the infarcted myocardium at 3 days and becomes more apparent in the following 2 weeks. Detected by a-SMA labeling, infarcted myocardium remains vascularized over 8 weeks (Fig. 1). Newly formed blood vessels accompany and nourish myoFb at the site of MI. 5. Myofibroblast activity and regulatory signals for tissue repair 5.1. Angiotensin II In addition to its well-described circulating endocrine properties, there is now accumulating evidence that AngII has important autocrine / paracrine functions in a variety of tissues [26,27]. The involvement of local AngII in tissue repair and fibrogenesis that follows inflammation has been inferred from experimental studies of MI. Local AngII peptide generation requires: the presence of requisite AngII peptide precursor, angiotensinogen (Ao); renin or cathepsin D that converts Ao to AngI; ACE, a membrane-bound ectoenzyme that provides extracellular hydrolysis of AngI to AngII. Ao is the only known precursor to Ang peptides and it is obligatory to tissue Ang generation and requires demonstration of Ao synthesis. Ao synthesis is present in rat and human cardiac tissue. In situ hybridization localizes Ao mRNA within fibroblasts and brown adipocytes [28,29]. Ao mRNA expression is found enhanced in the infarcted rat heart on day 5 after coronary artery ligation [30]. This precedes the morphologic appearance of fibrillar collagen in the form of fibrous tissue at this remote site. Renin synthesis is demonstrated in cultured fibroblasts and myocytes in neonatal rat hearts, but is very low in adult rat cardiac tissue. In situ hybridization reveals several fold increases in renin mRNA in the infarcted area [31]. Renin activity is also significantly increased in the infarcted myocardium 10 days after MI [32]. Various proteases, produced by the cell or procured from its environment, may also be involved in the generation of Ang peptides. These include cathepsin D and G, and other serine proteases that generate AngII directly from Ao [33,34]. 253 ACE mRNA and activity have been demonstrated in the heart of different species [35,36]. Localization and binding density of ACE in the normal and infarcted heart has been determined by in vitro quantitative autoradiography. ACE is heterogeneously distributed in the normal rat heart. Low-density ACE binding is found throughout ventricular myocardium and atria, whereas high-density binding is present at sites of high collagen turnover, including heart valve leaflets and the adventitia of intramyocardial coronary arteries [37]. Immunolabeling with a monoclonal ACE antibody identified cells expressing ACE. They include: endothelial cells on the surface of each valve leaflet; valvular interstitial cells residing within leaflet matrix; and fibroblast-like cells in the adventitia of intramural vessels, where they are responsible for collagen formation. High-density autoradiographic ACE binding is found at the site of MI at week 1 and increases progressively over the course of 8 weeks (Fig. 1) in parallel with morphological evidence of fibrillar collagen accumulation [10]. Increased ACE binding density in the infarct scar remains for at least 6 months, suggesting continued AngII generation at this site. ACE activity, as measured by substrate conversion, is increased in the infarcted myocardium, as is also the case for ACE activity at sites remote from the MI [10,38]. The concentration of AngII at the MI site is enhanced several fold [39]. Several cell types have been demonstrated to express ACE at the site of infarction. These include macrophages, endothelial cells, and myoFb [16]. Endothelial ACE is well positioned for circulating AngII generation, while ACE in macrophages and myoFb contributes to local AngII generation. Circulating renin– angiotensin–aldosterone system (RAAS) is, however, not activated in rats with MI [36,40], implicating the rise in AngII generation in the repairing myocardium is independent of circulating RAAS. Receptor–ligand binding is a requisite if locally generated AngII is to influence fibrogenesis. AngII receptors can be divided into two subtypes, AT1 and AT2. By autoradiography, atria and ventricles have been demonstrated to express low AngII receptors, while heart valve leaflets and aorta contain higher amounts of AngII receptors [41]. In the infarcted rat heart, high density AngII receptor binding is present at sites of repair over the course of 8 weeks following MI (Fig. 1) [42]. The specific AngII receptor subtype in the repairing rat myocardium is predominantly AT1. MyoFb are the primary contributor to AT1 receptor expression in the infarct scar [43]. These autoradiographic findings are consistent with the increase in mRNA and protein expression of the AT1 receptor found in homogenized tissue of the infarcted rat heart. The anatomic association between ACE and AT1 receptors at infarct scar raises the prospect that local concentration of AngII contributes to fibrous tissue formation. Campbell and Katwa [44] have reported AngII induced expression (mRNA and protein) of TGF-b1 by cultured myoFb mediated primarily by AT1 receptor-binding tissue. In vivo 254 Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 studies further reveal that AngII is correlated with TGF-b1 expression in repairing tissues, including infarcted heart and injured kidneys [22,44,45], suggesting AngII stimulates fibrous tissue formation by promoting TGF-b1 synthesis via AT1 receptor binding. Studies have demonstrated that in addition to collagen synthesis, AngII may regulate collagen degradation by attenuating MMP-1 activity [46] and enhancing TIMP-1 production [47], which further serve to promote collagen accumulation [48–50]. AT1 receptors are the predominant subtype expressed in the infarcted rat heart [43]. In the failing human heart, AT2 receptors are upregulated and fibroblast-like cells are responsible for their expression. AT2 receptors have therefore been linked to fibrosis, but this remains unclear at present. High-density ACE binding is also observed in endocardial and pericardial fibrosis that appear in the infarcted rat heart, as well as the pericardial fibrosis following sham operation (without MI). It also holds true for the foreign body fibrosis that surrounds the silk ligature placed around the left coronary artery and the infarcted rat kidney [51]. These findings strongly suggest AngII is involved in tissue repair irrespective of the etiologic basis of injury or the tissue involved. It further sheds light on why tissue ACE activity is increased in the infarcted heart and why AngII concentration is markedly increased at the site of MI. Both are a result of fibrous tissue and its cellular population. soon after induction of MI in rats or dogs, infarct size, hydroxyproline concentration of scar tissue, and myocardium bordering on the infarct were each reduced by these agents [53–57]. They likewise attenuated fibrous tissue formation at remote sites, e.g. interventricular septum and right ventricle, endocardium and visceral pericardium. In association with these interventions has been the attenuation in infarct tissue AngII concentration and TGF-b expression. Elevations in circulating renin, AngII and ACE are not observed in rats following MI, suggesting that locally produced AngII contributes to fibrogenesis in the repairing heart. The ability of these agents to protect an injured organ against unwanted fibrosis, mediated by the expression and elaboration of AngII and TGF-b1, has now been demonstrated in multiple organs after diverse forms of injury, including kidney, lung, liver and skin. Findings from multiple laboratories whose research is focused on addressing the regulation of unwanted fibrous tissue formation have underscored the importance of de novo generation of AngII by myoFb and autocrine induction of the fibrogenic cytokine TGF-b1 by this peptide in mediating tissue repair. This is now recognized as a common paradigm of repair in many injured organs. 6. Contractile behavior of infarct scar 5.2. Transforming growth factor-beta 1 TGF-b1 is an important regulatory peptide in fibrous tissue formation and has numerous actions on extracellular matrix. It stimulates fibroblast-like cell growth, enhances collagen synthesis, and suppresses collagen degradation [52]. By in situ hybridization, transcription of TGF-b1 mRNA increases at the site of MI soon after MI and remains elevated for many weeks (Fig. 1) [22]. The concentration of TGF-b1 is also increased in the infarcted rat myocardium week 4 following MI, implicating TGF-b1 synthesis in the infarct scar [22]. Cells accountable for TGF-b1 synthesis in the infarcted heart are primarily macrophages in the early phase of repair and myoFb in the fibrogenic phase of healing. The cellular actions of TGFb1 are mediated by its specific membrane-bound receptors. By in vitro autoradiography, TGF-b receptor binding density is found upregulated in the infarcted heart and remains so for weeks [22]. 5.3. Pharmacologic interventions Pharmacologic interventions with either an ACE inhibitor or an AT1 receptor antagonist have further underscored the importance of locally generated AngII and TGF-b1 in promoting tissue remodeling. Introduced at or MyoFb contain a-SMA and have cell–cell connections via gap junctions and cell–matrix connections via a fibronexus. This provides for a contractile assembly that contributes to scar tissue remodeling [3]. Contractile myoFb remain abundant in the infarct scar for months (Fig. 1) and progressive infarct thinning occurs over the course of 8 weeks. Such fibrous tissue contraction is stimulated by various substances, including AngII, catecholamines and endothelin-1 [58]. 7. Summary Infarct scar, long considered inert, is a dynamic tissue: cellular, vascularized, metabolically active, and contractile. It is composed of myoFb, which express components requisite to Ang peptide generation, including ACE, AT1 receptors with regulating fibrogenic cytokine TGF-b1. MyoFb, nourished by a neovasculature are persistent in infarct scar, where they continue to elaborate fibrillar type I collagen. Their generation of these peptides contribute to ongoing scar tissue collagen turnover and to fibrous tissue formation with structural remodeling of noninfarcted sites remote to MI. Infarct scar contraction accounts for its thinning and its tonus may contribute to abnormal ventricular chamber stiffness and diastolic dysfunction. Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 References [1] Jugdutt BI, Amy RWM. Healing after myocardial infarction in the dog: changes in infarct hydroxyproline and topography. J Am Coll Cardiol 1986;7:91–102. [2] Jugdutt BI, Schwarz-Michorowski BL, Khan MI. Effect of longterm captopril therapy on left ventricular remodeling and function during healing of canine myocardial infarction. J Am Coll Cardiol 1992;19:713–721. [3] Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR, Majno G. Granulation tissue as a contractile organ. A study of structure and function. J Exp Med 1972;67:719–734. [4] Darby I, Skalli O, Gabbiani G. a-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest 1990;63:21–29. [5] Lipper S, Kahn LB, Reddick RL. The myofibroblast. Pathol Annu 1980;15:409–441. [6] Birkedal-Hansen H, Moore WGI, Bodden MK et al. Matrix metalloproteinases: A review. J Cell Biochem 1993;51:326–335. [7] Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol 1995;27:1281–1292. [8] Sun Y, Zhang JQ, Zhang J, Lamparter S. Cardiac remodeling by fibrous tissue following infarction in rats. J Lab Clin Med 2000 (in press). [9] Cleutjens JPM, Verluyten MJA, Smits JFM, Daemen MJAP. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 1995;147:325–338. [10] Sun Y, Cleutjens JPM, Diaz-Arias AA, Weber KT. Cardiac angiotensin converting enzyme and myocardial fibrosis in the rat. Cardiovasc Res 1994;28:1423–1432. [11] Whittaker P. Unravelling the mysteries of collagen and cicatrix after myocardial infarction. Cardiovasc Res 1996;31:19–27. [12] Volders PGA, Willems IEMG, Cleutjens JPM, Arends J-W, Havenith MG, Daemen MJAP. Interstitial collagen is increased in the noninfarcted human myocardium after myocardial infarction. J Mol Cell Cardiol 1993;25:1317–1323. [13] Van Krimpen C, Smits JFM, Cleutjens JPM et al. DNA synthesis in the non-infarcted cardiac interstitium after left coronary artery ligation in the rat heart: effects of captopril. J Mol Cell Cardiol 1991;23:1245–1253. [14] Litwin SE, Litwin GM, Raya TE, Warner AL, Goldman S. Contractility and stiffness of noninfarcted myocardium after coronary ligation in rats: effects of chronic angiotensin converting enzyme inhibition. Circulation 1991;83:1028–1037. [15] Pelouch V, Dixon IMC, Sethi R, Dhalla NS. Alteration of lagenous protein profile in congestive heart failure second myocardial infarction. Mol Cell Biochem 1993;129:121–131. [16] Sun Y, Weber KT. Angiotensin converting enzyme and myofibroblasts during tissue repair in the rat heart. J Mol Cell Cardiol 1996;28:851–858. [17] Vracko R, Thorning D. Contractile cells in rat myocardial tissue. Lab Invest 1991;65:214–227. [18] Sappino AP, Schurch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab Invest 1990;63:144–161. [19] Skalli O, Schurch W, Seemayer T et al. Myofibroblasts from diverse pathologic settings are heterogeneous in their content of actin isoforms and intermediate filament proteins. Lab Invest 1989;60:275–285. [20] Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transform growth factor-b1 induces a-smooth muscle actin expression granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 1993;122:103–111. [21] Willems IEMG, Havenith MG, De Mey JGR, Daemen MJAP. The a-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol 1994;145:868–875. 255 [22] Sun Y, Zhang JQ, Zhang J, Ramires FJA. Angiotensin II transforming growth factor-b1 and repair in the infarcted heart. J Mol Cell Cardiol 1998;30:1559–1569. [23] Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995;146:56–66. [24] Zhang G, Moorhead PJ, El Nahas AM. Myofibroblasts and the progression of experimental glomerulonephritis. Exp Nephrol 1995;3:308–318. [25] Goumenos DS, Brown CB, Shortland J, El Nahas AM. Myofibroblasts, predictors of progression of mesangial IgA nephropathy. Nephrol Dial Transplant 1994;9:1418–1425. [26] Johnston CI. Biochemistry and pharmacology of the renin–angiotensin system. Drugs 1990;39:21–31. [27] Dzau VJ. Vascular renin–angiotensin system and vascular protection. J Cardiovasc Pharmacol 1993;22:S1–S9. [28] Campbell DJ, Habener JF. Cellular localization of angiotensinogen gene expression in brown adipose tissue and mesentery: Quantification of messenger ribonucleic acid abundance using hybridization in situ. Endocrinology 1987;121:1616–1626. [29] Cassis LA, Lynch KR, Peach MJJ. Localization of angiotensinogen messenger RNA in rat aorta. Circ Res 1988;62:1259–1262. [30] Lindpaintner K, Lu W, Niedermajer J. Selective activation of cardiac angiotensinogen gene expression in post-infarction ventricular remodeling in the rat. J Mol Cell Cardiol 1993;25:133–143. [31] Passier RCJJ, Smits JFM, Verluyten MJA, Daemen MJAP. Expression and localization renin and angiotensinogen in rat heart after myocardial infarction. Am J Physiol 1996;271:H1040–H1048. [32] Hirsch AT, Opsahl JA, Lunzer MM, Katz SA. Active renin and angiotensinogen in cardiac interstitial fluid after myocardial infarction. Am J Physiol 1999;276:H1818–1826. [33] Katwa LC, Tyagi SC, Campbell SE, Lee SJ, Cicila GT, Weber KT. Valvular interstitial cells express angiotensinogen, cathepsin D and generate angiotensin peptides. Int J Biochem Cell Biol 1996;28:807–821. [34] Katwa LC, Campbell SE, Tyagi SC, Lee SJ, Cicila GT, Weber KT. Cultured myofibroblasts generate angiotensin peptides de novo. J Mol Cell Cardiol 1997;29:1375–1386. [35] Dzau VJ. Cardiac renin–angiotensin system. Am J Med 1988;84:22– 27. [36] Hirsch AT, Talsness CE, Schunkert H, Paul M, Dzau VJ. Tissuespecific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circ Res 1991;69:475–482. [37] Yamada H, Mendelsohn FAO. Localization of angiotensin converting enzyme in rat heart. Circ Res 1991;68:141–149. [38] Hokimoto S, Yasure R, Yasue H, Fujimoto K, Sakata R, Miyamoto E. Increased angiotensin converting enzyme activity in left ventricular aneurysm of patients after myocardial infarction. Cardiovasc Res 1995;29:664–669. [39] Yamagishi H, Kim S, Nishikimi T, Takeuchi K, Takeda T. Contribution of cardiac renin–angiotensin system to ventricular remodeling in myocardial-infarcted rats. J Mol Cell Cardiol 1993;25:1369– 1380. [40] Hodsman GP, Kohauki M, Howes LG, Johnston CI. Neurohumoral responses to chronic myocardial infarction in rats. Circulation 1988;78:376–381. [41] Allen AM, Yamada H, Mendelsohn FAO. In vitro autoradiographic localization of binding to angiotensin receptors in the rat heart. Int J Cardiol 1990;28:25–33. [42] Sun Y, Weber KT. Angiotensin II receptor binding following myocardial infarction in the rat. Cardiovasc Res 1994;28:1623– 1628. [43] Sun Y, Weber KT. Cells expressing angiotensin II receptors in fibrous tissue of rat heart. Cardiovasc Res 1996;31:518–525. [44] Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-b1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 1997;29:1947–1958. 256 Y. Sun, K.T. Weber / Cardiovascular Research 46 (2000) 250 – 256 [45] Pimentel Jr. JL, Sundell CL, Wang S, Kopp JB, Montero A, Martinez-Maldonado M. Role of angiotensin II in the expression and regulation of transforming growth factor-b in obstructive nephropathy. Kidney Int 1995;48:1233–1246. [46] Funck RC, Wilke A, Rupp H, Brilla CG. Regulation and role of myocardial collagen matrix remodeling in hypertensive heart disease. Adv Exp Med Biol 1997;432:35–44. [47] Chua CC, Hamdy RC, Chua BHL. Angiotensin II induces TIMP-1 production in rat heart endothelial cells. Biochim Biophys Acta 1996;1311:175–180. [48] Cleutjens JPM, Blankesteijn WM, Daemen MJAP, Smits JFM. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res 1999;44:232–241. [49] Unger T, Culman J, Gohlke P. Angiotensin II receptor blockade and end-organ protection: pharmacological rationale and evidence. J Hypertens Suppl 1998;16:S3–S9. [50] Tsutsumi Y, Matsubara H, Ohkubo N et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circ Res 1998;83:1035–1046. [51] Sun Y, Weber KT. Angiotensin converting enzyme and wound healing in diverse tissues of the rat. J Lab Clin Med 1996;127:94– 101. [52] O’Kane S, Ferguson MW. Transforming growth factor-bs and wound healing. Int J Biochem Cell Biol 1997;29:63–78. [53] Smits JFM, van Krimpen C, Schoemaker RG, Cleutjens JPM, Daemen MJAP. Angiotensin II receptor blockade after myocardial infarction in rats: effects on hemodynamics, myocardial DNA synthesis, and interstitial collagen content. J Cardiovasc Pharmacol 1992;20:772–778. [54] Michel JB, Lattion AL, Salzmann JL et al. Hormonal and cardiac effects of converting enzyme inhibition in rat myocardial infarction. Circ Res 1988;62:641–651. [55] De Carvalho Frimm C, Sun Y, Weber KT. Angiotensin II receptor blockade and myocardial fibrosis of the infarcted rat heart. J Lab Clin Med 1997;129:439–446. [56] Jugdutt BI, Khan MI, Judutt SJ, Blinston GE. Effect of enalapril on ventricular remodeling and function during healing after anterior myocardial infarction in the dog. Circulation 1995;91:802–812. [57] Schieffer B, Wirger A, Meybrunn M et al. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type I receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994;88:2273–2282. [58] De Mey JGR, Fazzi GE. A smooth muscle-like component in rat myocardial infarcts [abstract]. Hypertension 1996;28:696.