Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
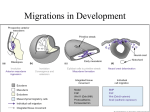
1 Developmental Biology and Morphogenesis of the Face, Lip and Palate Alphonse R. Burdi Prologue. The “history of man for the nine months preceding his birth would, probably, be far more interesting and contain events of greater moment than all the three score and ten years that follow it.” Samuel Taylor Coleridge, Miscellanies, Aesthetic and Literary. Circa 1800. The developmental biology of the face, lip, and palate is best understood against a backdrop of biological paradigms and information drawn from the multidisciplinary worlds of classical embryology, developmental biology, and, today, from the exciting world of molecular biology. The advent of many new and exciting clinical interventional strategies for the treatment of birth defects now allows the clinician to treat the most delicate of craniofacial abnormalities which were beyond the realm of treatment even for the skillful clinician due to lack of appropriate technologies. Even the details of craniofacial morphogenesis, however interesting to the clinician, were also seen as being beyond the everyday clinical realm, or esoteric, prior to the advent of the new wave clinical interventional techniques, such as high resolution imaging and information technologies, in the fields of craniofacial and maxillofacial surgery. Today, there is a rekindled need to understand more of the details of craniofacial morphogenesis, especially as that understanding increases our awareness of the etiology, pathogenesis, and clinical features of a variety of craniofacial defects. The goal of this chapter is to provide a highlighted update or working understanding of the “developmental blueprint” followed in human craniofacial morphogenesis, with a special focus on defects of the face, lip, palate, and associated structures. While recent advances in developmental and molecular craniofacial biology contribute heavily to the picture of face and palate morphogenesis, there has been an explosion in our fundamental understanding of the very beginnings of these body regions [43]. Such new information clearly centers on the genesis, behavior, and developmental outcomes of many craniofacial “building block “ cell types throughout their life span. These fundamental phenomena include patterns of early DNA signaling, gene and biochemical organizers, nuclear and cellular differentiation, proliferation, migration, and, importantly, patterns of inter- active behaviors at intracellular, cell surface, and extracellular matrix levels. Complete or partial interruptions of any one or combination of these phenomena have been implicated in the identification of etiologic and pathogenic causes of mammalian birth defects, including those of the human craniofacial regions. Normal and abnormal morphogenesis of the craniofacial regions, and even that of rest of the body, is dependent upon a myriad of cell types and tissues. One of the most important cell types in understanding normal and abnormal craniofacial morphogenesis is the neural crest cell [2, 36].While the importance of crest cells has been hypothesized for a century or more, not until the advent of neural crest biological markers – first with isotopic labels and later with specific markers as monoclonal antibodies, intracellular dyes, and protein assays – did the neural crest become so widely appreciated in a variety of studies of vertebrate embryogenesis in general, and human craniofacial morphogenesis, in specific. While the majority of recent neural crest studies of have necessarily dealt with chick and mouse embryos, there is ample evidence to show that basic information and associated technologies gained from vertebrate embryos can be directly applied to neural crest cells in mammalian and human embryos. With this in mind, prime consideration should be given to neural crest cells because they contribute heavily to craniofacial morphogenesis. These important “building-block” cells arise from the final stages in formation of the embryonic neural tube. Neural crest cell specificity is the result of an inductive action by nonneural ectoderm adjacent to the developing neural tube (mediated by bone morphogenetic proteins BMP-4 and BMP-7) on the lateral cells of the neural plate as the plate transforms from a plate of ectoderm into the definitive neural tube. The induced neural crest cells express the transcription factor slug which characterizes cells which separate from an em- 4 A. R. Burdi bryonic epithelial layer and subsequently migrate as mesenchymal cells away from the parent site [33]. The identification of the exact molecular mechanisms and cellular events linked to the differentiation, proliferation and, and especially, of the migration of crest cells into the facial and pharyngeal regions is not yet fully known. What is known, however, is that, with the variant OTX2 transcription factor, specific patterns of neural crest proliferation and migration into the pharyngeal arches are controlled by four members of homeodomain proteins called HOX genes (A-D). Another factor thought to be critical in the migration of crest cells is a loss of cell-to-cell adhesiveness which is associated with the loss of cell adhesion molecules (CAMs) characteristic of the neural tube and the migrating neural crest cells [91]. Following the completion of craniofacial crest cell migrations and differentiation into specific structures (such as bones of the facial skeleton) CAMs are re-expressed. Migrating craniofacial crest cells are thought to travel through cell-free intercellular spaces and pathways that have high levels of extracellular matrix molecules [3, 9, 10]. These migrations are determined by factors intrinsic to the crest cells themselves and features of the external environment through which the crest cells migrate. While most available information on neural crest migratory pathways comes from chick studies, data suggest that such information is applicable to mammalian and humans as well. Migrations are facilitated by the presence of such molecular substrates as fibronectin, laminin, and type IV collagen. Attachment to and migration of crest cells is mediated by a family of attachment proteins called integrins. It is important to note that other extracellular matrix molecules in the pathway (e.g., chondroitin sulfate-rich proteoglycans) can impede or block the normal migration of neural crest cells, which may lead to a number of craniofacial malformations. As will be expanded upon later in this chapter, neural crest cells have the remarkable capacity to differentiate into a wide variety of anatomical structures throughout the body. Exactly what controls crest differentiation is still an open question. One hypothesis is that all neural crest cells are equal in their developmental potential, and their ultimate differentiation is entirely predetermined by the environment through which the crest cells migrate and finally reside, i.e., “extrinsic determinants.” A second hypothesis favors “intrinsic determinants” and suggests that premigratory crest cells are intrinsically programmed for different developmental fates. Recent studies indicate that both hypotheses may be operative [1].While neural crest cells have a common site of origin during neural tube formation, not all neural crest cells behave alike. There are two families of crest cells, i.e., cranial and trunk neural crest. Trunk neural crest cells extend from lower cervical to the most caudal embryonic somites in humans. Trunk neural crest cells appear to have lessened migratory pathways and have fewer developmental outcomes than cranial neural crest cells, including formation of spinal ganglia, sympathetic ganglia, and adrenal medulla chromaffin cells. Interestingly, unlike cranial crest cells, trunk neural crest cells do not have the capacity to differentiate into skeletal tissues. The life history of cranial neural crest cells, while not of any more importance than trunk neural crest cells, appears to be more complex. Cranial neural crest cells are a major component of the embryo’s cephalic end and differentiate into a wide variety of cell and tissue types, including connective, skeletal, dentin, and muscle tissues of the face [64]. Unlike trunk neural crest cells, which show diffuse migratory pathways, cranial neural crest cells follow specific migratory pathways into specific regions of the embryonic head. They arise from the more cephalic neural tube regions and migrate ventrally into the pharyngeal arches adjacent to the upper regions of the embryonic gut tube. Such migrations are extensive and follow very definite migratory paths away from the neural tube and into the facial and pharyngeal regions. In hindbrain regions, neural crest cells arise from eight segmented regions on either side of the hindbrain (rhombencephalon) called rhombomeres (numbered R1-R8) and subsequently migrate into specific pharyngeal arches [12, 14]. Crest cells from R1 and R2 centers migrate into the first pharyngeal arch and play important roles in the formation of Meckel’s cartilage and the malleus and incus ear ossicles developing from it. Crest cells from R4 migrate into the second arch and contribute to the formation of the stapes, styloid process, and lesser horn of the hyoid bone. Crest cells from R6 and R7 migrate into the third arch, and those from R8 migrate into the fourth and sixth pharyngeal arches. The one variant noted above is that the first pharyngeal arch also includes crest cells from midbrain levels which express OTX2 transcription factors. Little evidence exists to show that crest cells from rhombomeric centers R3 and R5 play any significant role in human craniofacial morphogenesis. Crest cells initially express the HOX genes from their originating rhombomeric center, but maintenance of a specific expression is dependent upon interaction of the crest cells with the arch-specific mesoderm in the pharyngeal arches. While there is a specific linkage between given HOX genes and pharyngeal arches, morphologic derivatives arising from these linkages are also dependent upon epithelial-mesenchymal interactions and includes molecular signaling from surface ectoderm covering the arches, specifically, fibroblast growth factors (FGFs) which interact with underlying mes- Chapter 1 enchymal cells. Crest cells alone do not establish or maintain a specific pattern of morphologic expression. Several regulatory factors have been identified. Sonic hedgehog (Shh) genes and some retinoids have been shown to regulate normal HOX gene expression within the pharyngeal arches associated with a variety of developmental events, including neural plate development and craniocaudal body pattern formation. Defective differentiation, proliferation, and migration of cranial neural crest have been linked with a variety of developmental defects, i.e., the so-called neurocristopathies [24, 28, 29, 34]. Deficiencies and excesses of retinoids, for example, can disrupt proliferation and migration on specific crest cells, resulting in craniofacial defects, e.g., clefts of the lip and palate [54, 62]. With reference to the structural abnormalities in the chromosome 22-deletion DiGeorge syndrome, the fundamental pathogenesis for this clinical syndrome has been linked with defects in cranial neural crest cells of the third and fourth pharyngeal arches and cardiac outflow tract. Other cranial neurocristopathies include the wide range of craniofacial abnormalities in the frontonasal dysplasia family, Treacher Collins syndrome (mandibulofacial dysostosis), the Robin sequence, Waardenburg syndrome (types I and III), and neurofibromatosis (von Recklinghausen’s disease). The “building block “ cells for the head and face are identifiable both premorphologically and morphologically as early as the second intrauterine week. Once mapped out these cells continue with their peak period of cell differentiation, proliferation, and migration through the second intrauterine month.While the classical picture of craniofacial morphogenesis can be framed upon the morphogenesis of primary germ layer cells (i.e., ectoderm, mesoderm, endoderm), there is little doubt whatsoever that the current understandings and excitement about mammalian, including human craniofacial, morphogenesis have been significantly advanced by a plethora of studies of the origins and behavior of embryonic neural crest cells. Morphogenesis of the facial regions depends heavily on the timely differentiation, directed migration, selective proliferation of these crest cells which arise as a product of neural tube formation as the neural tube progressively pinches off from the overlying skin along the body’s dorsal axis. As will be discussed later, cells and tissues within each of the embryonic facial primordia arise from neural crest cells begin their migration (at about 21 postconception days) into the facial regions, as cell clusters called rhombomeres, from their sites of origin along the portions of the neural tube which form the brain. The determinants of crest cell migrations have been variously hypothesized as including intrinsic cell “targeting” factors and Developmental Biology and Morphogenesis chemical signaling from cells lining the extracellular cleavage planes through which the crest cells migrate [37, 42]. Crest cells from the developing midbrain regions migrate into upper facial regions, whereas crest cells from hindbrain migrate selectively into the lower facial regions [45–47]. Importantly, once the crest cells migrate into specific facial regions, they differentiate into mesenchymal cells that subsequently give rise to connective tissue and muscle cells of those specific facial regions [41]. While the predominant neural crest-derived mesenchymal cells in the facial regions do co-mingle with mesodermally derived mesenchymal cells, the interactive nature of their co-mingling, or lack thereof, remains uncertain. Consistent with the tenets of the “developmental field concept” [48] in human morphogenesis, both human and experimental studies generally have hypothesized that significant and early interference with normal differentiation, proliferation, and migration embryonic cells, including especially the craniofacial neural crest cells, can lead to isolated and syndromic craniofacial defects, called neurocristopathies, whose occurrence and severity depend on a combination of environmental and genotypic factors specific to a given dysmorphic or phenotypic trait [46, 47, 52]. Having addressed the general developmental features of the craniofacial “building block” neural crest cells, let’s turn our specific attention now to the key events in the shaping of the human face, lip, and palate. When the embryo’s cephalo-caudal axis is established at about 14 postconception days, the facial developmental field is one of the first of the head regions to appear [48]. Centrally-located in this region is a discrete bilaminar tissue plate, called the oropharyngeal membrane, whose structure and location marks the junction between the oral ectoderm and the endodermal digestive tube. This membrane progressively degenerates through the normal process of apoptosis or “programmed cell death” which involves increased phagocytic or lysosomal activity along the inner and outer surfaces of the membrane. Once the apoptosis of the oropharyngeal membrane is completed at 4 weeks, there is direct continuity between the spaces of the early oral cavity and the pharyngeal regions of the digestive tube. Only rarely does the oropharyngeal membrane fail to degenerate. Interestingly, a similar ecto-endodermal membrane lies at the depth of a groove which separates the first pharyngeal from the second pharyngeal arch. As will be discussed later, this membrane will have a very different and important developmental fate (i.e., does not undergo apoptosis) than that of the oropharyngeal membrane related to the fact that it has, unlike the oropharyngeal membrane, a layer of mesenchyme interposed between its ecto- and endodermal layers. 5 6 A. R. Burdi Clearly as much developmental “shaping” occurs on the laterals aspects of the young embryo’s head as in its the frontal regions. At four weeks, a series of lateral surface elevations, called pharyngeal arches, becomes quite prominent on the lateral side of the head. In fact, the appearance of the embryo’s head region at this time closely resembles the gill slit anatomy seen in a comparably-staged fish embryo; however, unlike in the fishes, the surface gill appearance in human embryos is short-lived, except as noted above, in the case of the development of the ear drum (or tympanum). The pharyngeal arches contribute significantly to the formation of the face, palate, and associated structures. Most congenital malformations of the head and neck have their beginnings during the cellular transformation of the pharyngeal arches into their adult derivatives. For example, branchial cysts and fistulae can occur in those rare instances in which human pharyngeal (or gill) clefts fail to smooth over on the lateral side of the neck. As mentioned earlier, cell masses which contribute to the bulging prominence of the arches are the neural crest cells that have migrated into the pharyngeal arches from specific brain regions, and which eventually differentiate into mesenchymal cells and give rise skeletal and muscular structures specific to a given pharyngeal arch. The first pair of pharyngeal arches are most important in shaping the human face and associated structures and will receive most attention in this chapter. The first pharyngeal arch, often called the mandibular arch, develops as two elevations around the oral opening which was filled in earlier by the oropharyngeal membrane. The larger and lower regions of this arch form much of the mandibular anatomy and the malleus and incus middle ear ossicles, whereas the smaller and upper regions of the first arch on either side of the oral opening give rise to the anatomy of upper lip, teeth, maxilla, zygomatic bone, and squamous portions of the temporal bone. The second pharyngeal arch is located beneath the first arch and is often called the hyoid arch in that it contributes significantly to the formation of the hyoid bone and one of the three middle ear ossicles, called the stapes. These two pharyngeal arches, like each of the other four pharyngeal arches, are separated from each other by a surface pharyngeal l groove which grows inwardly to meet an endodermal-lined outpocketing from the developing pharyngeal region, i.e., the first pharyngeal pouch. As is the case with most pharyngeal grooves and pharyngeal pouches, the contact zone between a pharyngeal l groove and a pharyngeal pouch is a bilaminar plate of ectoderm and endoderm which eventually degenerates, again through the process of “programmed cell death” and increased phagocytic activity. In the case of the first arch, however, this bilaminar plate is separated by invading crest-derived mesenchymal cells which have been linked with the failure of that specific plate to degenerate and persist normally throughout life as the adult eardrum, or tympanum. The elevated margins around the first pharyngeal groove develop through the selective proliferation of mesenchymal cells beneath the skin into six separate mesenchymal swellings, called auricular hillocks. These auricular hillocks progressively (from both the first and second pharyngeal arches) enlarge, migrate, and consolidate through programmed cell activity and eventually give rise to the external ear, or auricle. Failure of the auricular hillocks to develop normally can result in auricles of abnormal size, shape, and position as seen in a variety of isolated and syndromic craniofacial birth defects, e.g., first and second branchial arch syndrome, hemifacial microsomia, and microtia. The complete absence of the auricle (anotia) is a rare event. To complete this picture of the pharyngeal arches, it is important to note that cells within the arches are supplied by pairs of blood vessels, called aortic arches, that distribute blood from the embryonic heart upward through the tissue of each arch toward the brain and then down to the body [49]. As with the pharyngeal l arches themselves, not all of the aortic arches persist in humans. The aortic arches of the third, fourth, and sixth aortic arches do persist and become greatly modified throughout the embryonic period as they are reconstituted as the common carotid arteries which supply the neck, face, and brain. Especially important in this dynamic development of the craniofacial vasculature is the shifting of the primary arterial supply to the embryonic face prior to, during, and following the formation of the secondary palate. Unlike in the adult, prior to the seventh week, the primary source of blood to both the superficial and deep head tissues is the internal carotid artery and its branches. At about 7–8 weeks when the embryonic palatal shelves are experiencing their most critical stages of elevation and closure, an important shift occurs in the primary blood supply to the face and palatal tissues from the internal carotid to the external carotid arterial system. This transition involves a temporary vascular shunt between internal and external carotid systems provided by the stapedial artery. Failure of either the stapedial artery to form or failure of a complete and timely transition to occur has been hypothesized in identifying the pathogenesis of such conditions as palatal clefting and mandibulofacial dysostosis [51]. Considerably dependent upon the timely set morphogenic events that occur from the time of implantation through the fourth week, the embryonic face continues through its “developmental critical period,” which spans the fifth through seventh intrauterine weeks. It is that time period during which in human craniofacial morphogenesis generally is most suscep- Chapter 1 tible to either known or suspected birth defect-producing agents, or teratogens [66]. Arising from the first pharyngeal arch are four primordial or “building block” tissue masses that surround the large central depression of the primitive oral cavity. Continued morphogenesis of the facial prominences depends heavily upon the continuing migration, proliferation, and differentiation of the neural crest cells, under the direction of developmental morphogens, to a point in time when the facial prominences, or primordia, are clearly identifiable as the single median frontonasal prominence, paired maxillary prominences on either side of the frontonasal process, and two mandibular prominences beneath the oral opening. The shape and size of these prominences as well as development of the specific skeletal and muscular structures of each pharyngeal arch are critically dependent upon the continued viability and differentiation of the neural crest cells which are especially sensitive to teratogens, e.g., cortisone and retinoic acid [50, 61]. Continuing further with our focus on the developmental “blueprint” for the face, specifically the lip, it is important to note that the outcomes of several distinct brain-skin interactions in placode formation are also essential in early facial morphogenesis. By the beginning of the fifth week, oval patches of skin ectoderm lateral to the median frontonasal prominence interact with brain tissue to set off an ecto-ectodermal interaction resulting in the development of the two thickened nasal placodes located at the ventrolateral regions of the frontonasal prominence. Neural crestderived mesenchymal cells along the margins of the nasal placodes proliferate rapidly to produce horseshoe shaped elevations around the placode, called the medial and lateral nasal prominences, whose continued rapid growth gradually forms the nasal pits, or early nostrils. The forward growth of each lateral nasal process forms the ala of the nose, whereas the medial nasal process contributes to the formation of the nose tip, columella, the philtrum, tuberculum, and frenulum of the upper lip, and the entire primary palate. Through the process of relative growth in this area, the nasal placodes gradually “sink” to the depth of each nasal pit. Failure of the nose to develop completely is associated with failure of both nasal placodes to develop. A second important skin-brain interaction gives rise to localized thickenings of surface ectoderm on each side of the embryo’s head which will form the optic lens, retina, and nerve. Importantly, and as will be discussed later, these eye fields are first located on the lateral aspects of the embryo’s head and progressively migrate to the frontal midline at about the time the facial prominences are consolidating into the complete face [8]. Selective differentiation and proliferation of mesenchymal cells cause the maxillary prominences to Developmental Biology and Morphogenesis enlarge and migrate medially toward each other and the lateral and medial nasal prominences [12]. This migration is associated not only with patterns of cellular growth within the maxillary prominences, but also with timely migration of the eye fields from the lateral to the frontal regions of the embryo’s face during the fifth through eighth weeks [8]. Disturbances in normal eye field migration have been suggested as one possible cause of median facial clefting and the conditions of hypo- and hypertelorisms. Continued medial migration of the maxillary prominences on both sides also moves the medial nasal prominences toward the midline and each other. By the end of the sixth week, each maxillary prominence blends, or merges, with the lateral nasal prominence along a line which demarcates the future nasolacrimal groove and duct. This event then establishes the continuity between the side of the nose, or alar region, formed by the lateral nasal prominence with the cheek region formed from the maxillary prominence. A combination of reduced cell numbers and abnormal migration of mesenchymal cells can lead to the abnormal merging or consolidation of the maxillary and lateral nasal prominences. Although seen infrequently, this can lead to facial defects involving oblique facial clefts, persistent nasolacrimal grooves, and failure of the nasolacrimal duct to develop. Between the fourth and eight weeks, the medial nasal prominences merge with each other, small lower portions of the lateral nasal prominences, and with cells in the larger maxillary prominences. This subsurface merging of cells, especially between the medial nasal and maxillary prominences, results in the continuity of upper jaw and lip. As part of this consolidation of the medial nasal and maxillary prominences in upper lip formation, two important morphologic events need to occur. First, there is a deepening and downward growth of the nasal pit toward the oronasal cavity as a blind-ending sac whose floor eventually degenerates through programmedcell death resulting in the formation of the primitive choanae, which allows a continuity between the spaces of the primitive nasal cavity and the common oronasal cavity. An event occurring concomitantly with nasal pit morphogenesis is the formation of the seam between the intermaxillary segment and the maxillary prominence. As these two segments come together in the sixth week, the developmental surface seam of cells between them also elongates as the nasal pit elongates, deepens, and moves downward. This developmental seam, called the nasal fin, essentially forms the floor of the nasal pit and progressively degenerates by increased activity among phagocytic cells on either side of the seam [67]. Once “programmed cell” death of the nasal fin is essentially completed at about the seventh week, mesenchymal 7 8 A. R. Burdi cells from both the intermaxillary and maxillary prominences intermix, leading to fusion of the upper lip segments into the upper lip and its cupid’s bow. The completion of the embryonic lips generally occurs about 1 week earlier that the formation of the palate. Thus, the lips and palate have different “developmental critical periods” and, as such teratogens might affect either the lips or palate separately, or in combination. The intermixture of mesenchymal cells within the consolidated lip segments give rise to connective tissue components and muscle fibers within the orbicularis oris ring of the upper lip. Complete or incomplete failure of the nasal fin to degenerate have been associated with unilateral and bilateral clefts of the upper lip which variously involve abnormalities of the orbicularis oris muscle in terms of the numbers and distribution of its muscle fibers as part of the orbicularis ring. The incidence of orofacial clefts varies in accordance with the variances reported for differing population groups [22]. Examples of such population-specific differences, called polymorphisms, include cleft lip with or without cleft palate, which is one of the most common craniofacial birth defects in human with a reported incidence as high as 1:1000 in whites and higher in Asian and lower in black populations [13, 38, 56]. Another example of population polymorphisms shows that isolated cleft palate occurs more often in females (67%) than in age-matched males. This gender difference has been associated with a longer period of time for palatal closure in females which essentially increases the time during which female embryos might be affected by palatogenic teratogens [6]. Lateral clefts of the lip may or may not be associated with clefts of the palate. Mesenchymal cell deficiency that results in partial or complete failure of the two medial nasal prominences to consolidate into a philtrum can contribute to the formation of such defects as a bifid nose, or the rare median cleft (“hare lip”) of the upper lip, as characteristically seen in the autosomally recessive Mohr syndrome [23, 25, 30]. As the consolidation of facial processes progresses through the embryonic period, crest-derived mesenchymal cells within the maxillary prominences rapidly proliferate and differentiate into tissues which form mesenchymal cell fields from which the muscles of facial expression develop, and whose myofibers are innervated by the cranial nerve to the second arch, i.e., the facial nerve. Similarly, crest-derived mesenchyme in the maxillary and mandibular portions of the first pharyngeal arch differentiate predominately into the muscles of mastication, which are innervated by the trigeminal nerve of the first pharyngeal arch. Cells within the mandibular prominence give rise to muscle and connective tissue structures of the lower lip, chin, and lower cheek regions. With the reshaping and consolidation of the five major facial prominences, a recognizable human face is evident by the end of the eighth prenatal week [11, 40, 55, 58, 59]. Morphogenesis of the mammalian palate is an even more complex process which depends heavily upon the balance of genetic, hormonal, and various growth factors. As the face nears the completion of its “developmental critical period,” lateral palatine processes which form the secondary palate grow out from the walls of the still common oronasal cavity. The “developmental critical period” for the palate is from the end of the sixth week through the eighth intrauterine week, or 1 week longer in duration than that of the lip. These palatine shelves first grow medially, then become oriented inferolaterally to lie on either side of the tongue, which is quite precocious in its own development as a muscle-filled epithelial sac that fills much of the oronasal cavity. Nearing 8 weeks, the vertically oriented palatine shelves are progressively repositioned above the tongue mass. This repositioning of the shelves is thought to involve a combination of concurrent events, including a downward contraction of the tongue, an ameboid-like reshaping of the shelves which gradually places them over the tongue surface, an increase in extracellular shelf “forces” (or shelf fluid turgor) which reposition the shelves in a horizontal position, and a downward repositioning of the lower jaw [7, 18, 19]. In reality, normal or abnormal horizontalization of the palatine shelves is related to a combination of these three events. Palatal shelf elevation begins in the posterior regions of the shelves and depresses the tongue downward and forward and this allows the more anterior regions of the shelves to first contact one another near the posterior edge of the primary palate, or in the region of the future incisive canal [4]. Once the shelves are in a horizontal position, the shelves contact each other, and essentially stick together by a combination of interlocking shelf surface microvilli and a proteoglycans surface coating along the medial epithelial edge (MEE) of each shelf [39]. The elimination of the MEE is crucial for normal morphogenesis of the anterior regions of the secondary palate [15, 20, 26, 32]. Once the shelves make contact, there is a degeneration (i.e., apoptosis or programmed cell death) of epithelial cells along the abutting shelf linings, and a directed movement of crest-derived mesenchymal cells from one shelf to the other. This process of epithelial degeneration along with intershelf bridging of mesenchymal cells is called fusion. As is the case for craniofacial morphogenesis in general, several categories of factors (e.g., chromosomes, genes, signaling proteins, transcription factors, specific proteins) have either been identified or hypothesized as important in normal and abnormal palatogenesis [57, 64]. Fusion of the palatal shelves Chapter 1 has been linked to a variety of growth factors like those in the TGF-Beta-3 growth factor family [32, 53, 63] and protein activities [16, 31]. Complete or regional failures in programmed cell death MEE along the lengths of the palatal shelves can lead to various forms of palatal clefts.While great strides have been made in identifying genetic, cellular, and molecular controls of normal palatal development (mostly in mice and chick embryos), the identification of the exact balance between intrinsic and environmental controls of palatal morphogenesis remains elusive [44, 60]. The embryonic palatine raphe, or future midpalatine suture, marks the line of fusion between the palatine shelves. From the site of first shelf contact and fusion near the future incisive foramen, fusion of the more posterior regions of the shelves takes place over the next two weeks. Fusion also occurs between the shelves and the inferior edge of the nasal septum, except in the more posterior regions where the soft palate and uvula remain free. Once fusion of the shelves of the secondary palate is complete, their mesenchymal cells differentiate into osteogenic cells which form the skeletal elements of the premaxillary, maxillary, and palatine portions of the palate. Formation of the soft palate and uvula takes a slightly different course than that of the regions of the secondary palate which give rise to the hard palate [5]. The soft palate and uvula develop from two separate masses found at the most posterior portions of the secondary palatine shelves. Unlike the fusion mechanism which is in place along much of the length of the palatine shelves, the consolidation of these two separate masses is brought about by a selective proliferation of mesenchymal cells located deep in the valley between the masses. As that proliferation, called merging, continues the valley between the two distal shelf masses is obliterated, which results in a smoothening of the contour of the soft palate and uvula. Failure of the merging process in soft palate and uvula development can result in complete or partial clefts of the soft palate and uvula. Clefts of the palate, with or without clefts of the lip, are relatively common depending on the population group of the individual [23, 65]. Whereas occurrence figures for nonsyndromic cleft lips (with or without cleft palate) are about 1 in 1,000 live births, clefts of the palate (with or without cleft lip) occur in 1 in 2,500 live births, again depending on the population group of the individual, i.e., highest incidence in Asian, intermediate in white, and lowest in black populations [60]. Most clefts of the lip and palate generally are related to an interplay of genetic and environmental factors, i.e., multifactorial inheritance [21]. While animal studies have provided some insight into the molecular and cellular bases of these defects, precise etiologic explanations, especially involving teratogens in the Developmental Biology and Morphogenesis etiology of clefts of the human lip and palate, are still wanting. Crucial faulty chromosomes (see, for example, ref. 17) and genes linked to inherited forms of cleft lip and palate have recently been identified. This candidate gene is known as Interferon Regulatory Factor 6 [IRF6] [35]. Other candidate genes have been assigned some importance in palatal morphogenesis, including MSX1, LHX8 6p24 genes (associated with palatal shelf growth and differentiation); TGFA, EGFR and HOXA2 (associated with elevation and depression of tongue);TGFB3 and PVRL1 (associated with associated with sequential fusion stages along the midpalatal seam), and TGFA and EGFR (associated with actual apoptosis, or “programmed cell death” events along the midpalatal seam). Some clefts of the lip with or without cleft palate are seen regularly in a number of single mutant gene syndromes. Other clefts are associated with chromosomal syndromes, especially in trisomy 13. A complete cleft palate represents a maximum degree of clefting and is a birth defect in which the cleft extends from the incisive foramen region through the soft palate and uvula. The incisive foramen region is the demarcation used in distinguishing the two major groups of cleft lip and palate. Anterior cleft types include cleft lip, with or without a cleft of the alveolar region of the maxilla. A complete anterior cleft extends through the lip and alveolar region to the incisive foramen region. The pathogenesis of anterior clefts is related to a deficiency of neural crest-derived mesenchymal cells chiefly within the intermaxillary segment of the lip. The posterior cleft type of birth defect generally include clefts of the secondary palate that extend from the incisive foramen through the soft palate and uvula. The observation that the female secondary palate has a longer “developmental critical period” [6] than the male embryo by approximately 1 week offers some explanation why isolated cleft palate is more prevalent in females (66%) than males (34%). In general, the pathogenesis of posterior palatal clefts is related to abnormalities in a combination of events ranging from deficiencies in mesenchymal cell numbers to perturbations in the shelf extracellular matrices to abnormal elevation and fusion of the shelves, or lack thereof, as associated with a number of hypothesized teratogens, including excess doses of retinoic acids, glucocorticoids, and dioxins. 1.1 Summary The understanding of the natural history, clinical delineation, and clinical management of birth defects involving the face, lip, and palate has progressed significantly over the last 20 years and continues to do as we move further into the 21st century. Although hu- 9 10 A. R. Burdi man craniofacial morphogenesis is clearly the culmination of a very complex series of diverse and overlapping developmental events, all of these events can be categorized into four fundamental processes which span mammalian development and are evident in the earliest beginnings of the face and palate – normal and abnormal: (1) cell differentiation – the process through which the myriad of “building block” cell types invoked in facial morphogenesis are generated from the single-celled zygote; (2) morphogenesis – the process or set of processes through which the complex form of the face and its constituent cells, tissues, and organs will emerge in a timely fashion along patternable individual and population lines; (3) growth – the collective results of differentiation and morphogenesis; and (4) dysmorphogenesis and abnormal growth – this is the most exciting of the challenges we face today as we strive to understand how environmental influences interact with and cause changes in the expression of the genetic factors governing the behavior of those cells which will give rise to the entire human body, and especially the face and palatal regions. The treatment of defective genes is very much a part of the current clinical agenda dealing with craniofacial defects. The basic scientist, the dysmorphologist, the clinician, and, importantly, those with natural or acquired craniofacial defects have gained significant advantage from the critical use of available information coming from classical and experimental studies of human morphogenesis. These advantages will continue to increase as laboratory scientists and clinician scholars move rapidly together into the world of molecular and gene biology. These approaches should and will increase our knowledge base on the patterns and underlying causes of normal and abnormal craniofacial morphogenesis – and our patients will be all the better for it. However, most researchers and treatment providers well realize that the practical transfer of new biological information on normal and abnormal development flowing from the laboratory bench to the clinical bedside may be neither easy nor timely to achieve in the effective treatment and management of craniofacial abnormalities. Epilogue. “And the end of all of our exploring will be to arrive where we started and know the place for the first time.” T.S. Eliot, 1918 References The following references have been selected from an ever-growing literature in developmental biology and craniofacial biology in particular. Perusal of any one or several of these references should lead the reader to further readings which bear on the subject of the biology of face, lip and palate development – normal and abnormal. References noted with an (*) are texts containing illustrations that elucidate the information contained in this chapter. References 1. Anderson DJ. Cellular and molecular biology of neural crest lineage determination. Trends Genet 1997; 13:276– 280. 2. Birgbauer E, Sechrist J, Bronner-Fraser M, Fraser S. Rhombomeric origin and rostro caudal reassortment of neural crest cells revealed by intravital microscopy. Development 1995; 935–945. 3. Brinkley L, Morris-Wiman J. The role of extracellular matrices in palatal shelf closure. In: Zimmerman EF (ed) Current trends in developmental biology. New York: Academic Press; 1984. 4. Burdi AR, Faist K. Morphogenesis of the palate in normal human embryos with special emphasis on the mechanisms involved. Am J Anatomy 1967; 120:149–160. 5. Burdi AR. Distribution of midpalatine cysts: A re-evaluation of human palatal closure mechanism. J Oral Surg 1968; 26:41–45. 6. Burdi AR, Silvey R. Sexual differences in closure of the human palatal shelves. Cleft Palate J 1969. 6:1–7. 7. Burdi AR, Feingold M, Larsson KS, Leck I, Zimmerman E. Etiology and pathogenesis of congenital cleft lip and cleft palate. An NIDR State of the Art Report. Teratology 1972. 6:255–270. 8. Burdi AR, Lawton TJ, Grosslight J. Prenatal pattern emergence in early human facial development. Cleft Palate J 1988; 25:8–15. 9. Bronner-Fraser M. Experimental analyses of the migration and cell lineage of Avian neural crest cells. Cleft Palate J 1990; 27:110–120. 10. Bronner-Fraser M, Fraser SE. Cell lineage analysis of the avian neural crest. Development 1991; 2:17–22. 11.* Carlson BM. Human embryology & developmental biology. 2nd.ed. St. Louis: Mosby; 1999. 12. Carstens MH. Development of the facial midline. J Craniofacial Surg 2002; 13:129–187. 13. CDC National Center on Birth Defects and Developmental Disabilities. 2002. http://www.cdc.gov/ncbdd. 14. Couly G, LeDourain NM. Head morphogenesis in embryonic avian chimeras: evidence for a segmental pattern in the ectoderm corresponding to neuromeres. Development 1990; 108:543–558. 15. Cuervo R, Valencia C, Chandraratna RAS, Covarrubias L. Programmed cell death is required for palatal shelf fusion and is regulated by retinoic acid. Dev Biol 2002; 245:145– 156. 16. Darling DS, Stearman RP, Qiu M, Feller JP. Expression of Zfhep/EF1 protein in palate, neural progenitors, and differential neurons. Gene Expr Patterns 2003; 3:709–717. 17. Davies AF. Further evidence for the involvement of human chromosome 6p24 in the aetiology of orofacial clefting. J. Med Genetics 1998; 35:857–861. 18. Diewert VM. The role of craniofacial growth in palatal shelf elevation. In: Pratt RM, Christiansen RL (eds). Current research trends in prenatal craniofacial development. New York: Elsevier North-Holland; 1980. Chapter 1 19. Ferguson MWJ. Palatal development. Development 1988; 103:41–57. 20. Fitchett JE, Hay ED. Medial edge epithelium transforms to mesenchyme after palatal shelves fuse. Dev Biol 1989; 131: 455–474. 21. Fraser FC. The multifactorial threshold concept – uses and abuses. Teratology 1976 14:267–280. 22. Freni SC, Zapisek WF. Biologic basis for a risk assessment model for cleft palate. Cleft Palate J 1991; 28:338–346. 23. Gelehrter TD, Collins FS, Ginsberg D. Principles of medical genetics. 2nd ed. Baltimore: Williams & Wilkins; 1998. 24. Goodman RM, Gorlin RJ. The malformed infant and child. New York: Oxford University Press; 1983. 25. Gorlin RJ, Cohen MM Jr., Levin LS. (eds) Syndromes of the head and neck 4th ed. New York: Oxford University Press; 2002. 26. Griffith CM, Hay ED. Epithelial-mesenchymal transformation during palatal fusion: carboxyfluorescein traces cells at light and electronmicroscopic levels. Development 1992; 116:1087–1099. 27. Hunt P, Wilkinson D, Krumlauf R. Patterning the vertebrate head: murine Hox 2 genes mark distinct subpopulations of premigratory and migratory cranial neural crest. Development 1991; 112:43–50. 28. Johnston MC, Bronsky PT. Craniofacial embryogenesis: abnormal developmental mechanisms. Ch. 5:61–124. In: Mooney MP, Siegel ML, (eds) Understanding craniofacial anomalies. New York: Wiley and Liss; 2002. 29. Jones KL. Smith’s recognizable patterns of human malformation. 4th ed. Philadelphia: W.B. Saunders; 1990. 30. Jones M. The neurocristopathies: reinterpretation based on the mechanism of abnormal morphogenesis. Cleft Palate J 1990; 27:136–149. 31. Kang P, Svoboda KKH. PI-3 kinase activity is required for epithelial-mesenchymal transformation during palatal fusion. Dev Dynamics 2002; 225:316–321. 32. Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta 3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dynamics 1997; 209:255–260. 33. Keynes R, Cook GMW.Axon guidance molecules. Cell 1995; 83:161–169. 34. Kissel P, Andre JM, Jacquier A. The neurocristopathies New York: Masson; 1981. 35. Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, Ferreira de Lima RLL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in interferon regulatory factor 6 cause vander Woude and popliteal pterygium syndromes. Nat Genet 2002; 32:285–289. 36. Le Dourain N. The neural crest. Cambridge, England: Cambridge University Press; 1982. 37. Lumsden A, Sprawson N, Graham A. Segmental origin and migration of neural crest in the hindbrain region of the chick embryo. Development 1991; 113:1281–1291. 38. March of Dimes Birth Defects Foundation. 2002. http://www.modimes.org/. 39. Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O’Kane S, Puerta J, Ferguson MW. Medial edge epithelium cell fate during palatal fusion. Dev Biol 2000; 220:343–357. Developmental Biology and Morphogenesis 40. Moore K, Persaud TVN. The developing human. 6th ed. Philadelphia: W.B. Saunders; 1998. 41. Morris-Kay G, Tan S-S. Mapping neural crest cell migration pathways in mammalian embryos. Trends Genet 1987; 3:257–261. 42. Morris-Kay G, Tuckett F. Early events in mammalian craniofacial morphogenesis. J Craniofac Genet Dev Biol 1991; 11:181–191. 43. Nieto MA. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994; 264:835– 839. 44. Murray J. Gene/environmental causes of cleft lip and/or palate. Clin Genet 2002; 61:248–256. 45. Noden DM. Origins and patterning of craniofacial mesenchymal tissues. J Craniofac Genet Dev Biol 1986; 2:15–31. 46. Noden DM. Cell movements and control of patterned tissue assembly during craniofacial development. J Craniofac Genet Dev Biol 1991; 11:192-213. 47. Noden DM. Vertebrate craniofacial development: the relation between ontogenetic process and morphologic outcome. Brain Behav Evol 1991; 190–225. 48. Opitz JM. The developmental field concept New York: Liss; 1986. 49. Padget D. The development of the cranial arteries in the human embryo. Carnegie Contrib Embryol 1948; 32:205–268. 50. Persaud TVN. Environmental causes of human birth defects Springfield: Charles C Thomas; 1990. 51. Poswillo DE. The pathogenesis of the first and second branchial arch syndrome. Oral Surg, Oral Med, Oral Pathol 1973; 35:302–328. 52. Pratt RM. Environmental factors influencing craniofacial morphogenesis. In: Vig KWL, Burdi AR (eds). Craniofacial morphogenesis and dysmorphogenesis. Craniofacial growth series. Monograph 21. Ann Arbor: University of Michigan Center for Human Growth & Development; 1988. 53. Proetzel G, Pawlowski SA,Wiles MV,Yin M, Boivin GP, Howels PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 1995; 11:409–414. 54. Rowe A, Richman JM, Brickell PM. Retinoic acid treatment alters the distribution of retinoic acid receptors in the embryonic chick face. Development 1991; 111:1007–1016. 55. Sadler TW. Langman’s medical embryology. 9th ed. Baltimore: Lippincott Williams & Wilkins; 2004. 56. Schutte BC, Murray J. The many faces and factors of orofacial clefts. Hum Mol Genet 1999; 8:1853–1859. 57. Slavkin H. Regulatory issues during early craniofacial development. Cleft Palate J 1990; 27:101–109. 58. Sperber GH. Craniofacial embryology. 5th ed. Hamilton, Canada: BC Decker; 2001. 59. Sperber GH. First year of life: prenatal craniofacial development. Cleft Palate J 1990; 29:109–111. 60. Spritz RA. The genetics and epigenetics of orofacial clefts. Curr Opin Pediatr 2001; 13:556–560. 61. Sulik KK, Dehart DB. Retinoic-acid induced limb malformations resulting from apical ectodermal ridge cell death. Teratology 1988; 37:527–537. 62. Sulik KK, Johnston MC, Cook CS, Webster WS. Teratogensand craniofacial malformations. Dev Biol 1988; 103:213– 232 63. Taya Y, O’Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta 3 knockout mice. Development 1999; 126: 3869–3879. 11 12 A. R. Burdi 64. Thesleff I, Nieminen P. Molecular mechanisms of cell and tissue interactions during early tooth development. Anat Rec 1996; 245:151–161. 65. Thompson MW, McInnes RR, Willard HF. Thompson & Thompson genetics in medicine. 5th ed. Philadelphia: W.B. Saunders; 1991. 66. Vig KWL, Burdi AR. Craniofacial morphogenesis and dysmorphogenesis. Craniofacial Growth Series Monograph No.21. Ann Arbor; The University of Michigan Center for Human Growth & Development; 1988. 67. Warbrick JG. The early development of the nasal cavity and upper lip in the human embryo. J Anat 1960; 94:351–362.