Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Deoxyribozyme wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Biochemistry wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Microbial metabolism wikipedia , lookup
Sulfur cycle wikipedia , lookup
Biosynthesis wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Catalytic triad wikipedia , lookup
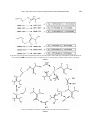
Indian Journal of Chemistry Vol. 50A, Sept-Oct 2011, pp. 1457-1462 Density functional theory calculations on biological S-transfer: Insight into the mechanism of rhodanese Subal Dey & Abhishek Dey* Department of Inorganic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India Email: [email protected] Received 8 June 2011; revised and accepted 10 August 2011 Biological sulfur transfer (S-transfer) is a key step in the synthesis of metabolites, CN- detoxification and assembly of iron-sulfur clusters. Computational results addressing the thermodynamics of the S-transfer reactions from thiosulfate (natural S-donor) to HCN and thiol are presented. These calculations indicate that S-transfer from thiosulfate to HCN and thiol is possible only in the anionic forms of these species. However these species have pKa values significantly higher than physiological pH value (i.e., they are protonated in physiological pH and incapable of S-transfer). In the rhodanese active site, basic residues are present to deprotonate the catalytic cysteine group which accepts the S-atom from thiosulfate. The resultant perthiol species transfer S-atom to CN- in a synchronous S-atom and H+ transfer step facilitated by the two arginine residues present in the rhodanese active site. Based on these calculations, a mechanism is proposed for the rhodanese catalyzed CN- detoxification pathway. Keywords: Density functional calculations, S-transfer reactions, Cyanide detoxification, Rhodanese The presence of sulfur in biological cofactors has been established since the early 20th century.1 Various vitamins, amino acids, iron-sulfur clusters, lipoic acid, biotin and thiouridine contain sulfur atom as functional motifs of these biologically important compounds.2 Photosynthetic sulfur bacteria use the sulfur compounds as a source of electrons for reductive carbon dioxide fixation during anoxic autotropic growth, while chemolithoautotropic sulfur bacteria uses the electron derived from oxidation of inorganic sulfur species both in carbon dioxide fixation and as respiratory electron donors.3,4 In the last decade great strides have been made towards understanding the biosynthesis of these processes. It has been established that the cystein desulfurase and the rhodanese homology domains are essential for catalyzing the insertion of the sulfur atoms into bimolecules.2,5 Remarkably, all the enzymes use perthiols as an intermediate for the sulfur transfer, when perthiols, as a chemical entity, is quite uncommon in synthetic preparation.6 Persulfides, on the other hand, are well characterized.7-9 Rhodanese an ubiquitous structural module occurring in three major evolutionary phyla, is the enzyme that can catalyse the conversion of thiosulfate to thiocyanide in the presence of cyanide, i.e., thiosulfate:cyanide sulfurtransferase(TST).10-12 Rhodanese from bovine13 and Azatobacter vinelandii (RhdA)14 is crystallographic characterized. It consists of two equally-sized globular domains:13 the active C-terminal domain which contains the active site loop hosting six amino acid residues with one cysteine residue at the first position and the catalytically inactive N-terminal domain where the Cys residue is replaced by an Asp residue.13,15-17 The peculiarity of rhodanese resides in two conserved patterns of amino acid sequences at the N-terminal region ([F/Y]-X3-H-[L/I/V]-P-G-A-X2-[L/I/V]) and at the C-terminal end of the protein ([A/V]-X2-[F/Y][D/E/A/P]-G-[G/S/A]-[W/F]-X-E-[F/Y/W]).18 The sulfur transfer catalysis at the rhodanese active site occurs via a proposed double displacement mechanism involving the formation of a transient cysteine persulfide intermediate,2 in which the transferring sulfur is bound to the catalytic Cys residue (Fig. 1). This sulfane sulfur (S0) is proposed to be nucleophilic in nature and easy to deliver to cyanide to form thiocyanide in the second step of the reaction.19-22 The catalytic cysteine sulfur is H-bonded to basic arginine residues which are also proposed to be mechanistically important. It may be assumed as the cyanide detoxification pathway within the cell and may appear as the major contributing component of the recovery mechanism for the native architecture of 1458 INDIAN J CHEM, SEC A, SEPT-OCT 2011 Table 1—Calculated solvation enthalpies Species Solvation enthalpy (kcal/mol) - CH3S CH3SH HSCN CNCH3SSH SCNCH3SSHCN S2O32SO32- Fig. 1—Active site of bovine rhodanese from (pdb id: 2ORA). The hydrogen bonding interaction with the catalytic Cys254 residue and Arg110 and Arg116 are indicated with dashed lines. the iron-sulfur protein(s) as it can mobilize sulfur for the formation or repair of iron-sulfur clusters.22,23 In spite of the importance of this reaction in biology, little is known about the thermodynamics of these reactions. In this paper, the feasibility of the S-transfer between several relevant species has been investigated by density functional theory calculations in the gas phase as well as using a solvation model. These calculations provide insight into the possible role and mechanism of rhodanese. Computational Details All calculations were performed in the HPC cluster at IACS, Kolkata, using Gaussian 03 software package. The geometries were optimized using spin restricted formalism using both BP8624,25 and B3LYP26-28 functional and a 6-311g* basis set. All coordinates were allowed to optimize. Frequency calculations at the end of geometry minimization were done to ensure a stable minimum. The optimized structures reported had no imaginary frequencies. The total energies were calculated using 6-311+g* basis set on all atoms. A polarized continuum model (PCM) using water as solvent was used to model solvation.29 Free energies differences (∆G) were calculated by correcting the differences in electronic energies (∆E) obtained using a 6-311+g* basis with the entropy (S) and zero point energies (ZPE) obtained during the frequency calculations using 6-311g* basis set. The results obtained using B3LYP and BP86 functional were qualitatively the same so only those obtained using BP86 is reported here. -65.226 -0.32484 -8.45525 -79.2471 -2.03738 -61.6453 -60.513 -4.08202 -224.733 -276.194 Results and Discussion Table 1 lists the calculated solvation energies for the species relevant to this study. The results are divided into two sections: Section A discusses the thermodynamics of S-transfer from thiosulfate. In this section thiosulfate is always assumed to be in its dianionic form as the pKa of H2S2O3 is 0.6 and 1.72 (ref. 30). Thus, thiosulfate should exist in its dianionic form in physiologically relevant pHs. In section B, S-transfer from perthiolate species is considered. In this section both the protonated and the deprotonated forms are considered since perthiol has a pKa of 6.2 ± 0.1 and it can exist in both neutral as well as anionic forms depending on the pH or the nature of neighboring residue (i.e., basic residues, like Arg116 can deprotonate a perthiol.).31 Transfer of S-atom along with a proton has also been considered as there is a conserved basic group in the rhodanese active site that can allow this synchronous shuttling. (A) S-transfer from thiosulfate to (i) Cyanide: S-transfer from thiosulfate to cyanide produces thiocyanide and sulfite: 2− − 2− − S2O3 + CN → SO3 + SCN . This reaction is calculated to be thermodynamically favorable in the gas phase (∆E = −4.514 kcal/mol, ∆G = −4.906 kcal/mol). This reflects differences in the bond strengths between the product and the reactant. In a PCM model using water as solvent, this reaction is calculated to be favorable (∆E = −18.504 kcal/mol, ∆G = −18.897 kcal/mol). The solvation of SO32− is greater than S2O32− by 52 kcal/mol (Table 1), while the solvation energy of SCN- is lower by 18 kcal/mol. Thus, the greater solvation of the product SO32provides the driving force for the reaction in an aqueous environment. There is minimal entropic contribution to the calculated ∆G of this reaction. DEY & DEY: DFT CALCULATIONS ON BIOLOGICAL S-TRANSFER REACTIONS (ii) Hydrocyanic acid: The sulfur transfer from thiosulfate to hydrocyanic acid will result in the formation of hydrothiocyanic acid and sulfite: HCN + S2O32− → HSCN + SO32−. The thermodynamic values (∆E = 68.934 kcal/mol and ∆G = 66.86 kcal/mol) indicate that the reaction is unfavorable. Here, proton affinity (PA) plays the leading role. The gas phase PA of CN− (−26.059 kcal/mol) is significantly greater than the gas phase PA of SCN− (1.473 kcal/mol). This makes the reaction thermodynamically unfavorable. Upon introducing solvation correction, the ∆G is lowered from +66.86 kcal/mol in the gas phase to 8.53 kcal/mol in water because of the larger solvation energy of SO32- (product) relative to that of S2O32− (reactant). However, the higher PA of HCN relative to the PA of HSCN disfavors the S- transfer from thiosulfate to hydrocyanic acid. (iii) Methylthiolate: S-transfer from thiosulfate to thiolate will yield sulfite and perthiolate: CH3S− + S2O32− → CH3SS− + SO32−. The reaction is calculated to be thermodynamically unfavorable in the gas phase (∆E = 51.71 kcal/mol and ∆G = 50.51 kcal/mol). This is probably due to weakening of the S−S bond in perthiolate species relative to that in thiosulfate. However, the reaction is calculated to be slightly thermodynamically favorable after solvation correction (∆E = −0.661 kcal/mol and ∆G = −1.86 kcal/mol). The solvation energy of the reactant methylthiolate (−65.23 kcal/mol) is ~5 kcal/mol more than the solvation energy of methylperthiolate (−60.51 kcal/mol). This will tend to make the ∆G of the reaction unfavorable. However the large solvation energy of SO32− overcomes this as well as the gas phase endothermicity to enable the reaction in the aqueous medium. (iv) Methylthiol: Methylthiol on reacting with thiosulfate will generate methylperthiol and sulfite: CH3SH + S2O32− → CH3SSH + SO32−. From the calculations we can conclude that this reaction is thermodynamically unfavorable in the gas phase as well as in the aqueous media. ∆E = 63.25 kcal/mol and ∆G = 62.28 kcal/mol in the gas phase imply that the S−S bond in perthiol is weaker than the S−S bond in thiosulfate. Further, the gas phase PA of methylthiol (−29.65 kcal/mol) is ~ 8 kcal/mol higher than the gas phase PA of methylperthiol (−21.83 kcal/mol). Both these factors make the reaction thermodynamically unfavorable. Although solvation correction favors this reaction (due to the large solvation energy of SO32−), it does not 1459 overcome the gas phase endothermicity of this reaction (∆G = 10.31 kcal/mol in water). (B) (I) S− transfer from CH3SS− to (i) Cyanide: Perthiolate to cyanide S-transfer will result in the formation of thiocyanate and thiolate: CH3SS− + CN− → CH3S− + SCN−. This reaction is calculated to be favorable in the gas phase as well as in water. In the gas phase, formation of stronger C−S bond in SCN− and breaking of weak S−S bond in perthiolate makes the reaction favorable (∆E = −35.668 kcal/mol and ∆G = −34.967 kcal/mol). Including solvation correction makes the reaction less thermodynamically favorable (∆E = 21.977 kcal/mol and ∆G = −21.276 kcal/mol). This is because the calculated solvation energy of the reactant CN− (−79.25 kcal/mol) is ~18 kcal/mol more negative than the calculated solvation energy of the product SCN(−61.64 kcal/mol). Thus, in an aqueous medium the reactant is stabilized relative to the product making the ∆G more positive. (ii) Hydrocyanic acid: Methyl perthiolate on reaction with hydrocyanic acid results in the formation of methyl thiolate and thiocyanic acid: CH3SS− + HCN → CH3S− + HSCN. The reaction is not favorable in the gas phase (∆E = 13.916 kcal/mol and ∆G = 13.086 kcal/mol) because the proton affinity of the reactant CN− (−26.68 kcal/mol) is much greater than that of the product, SCN− (1.47 kcal/mol). Though the solvation of the ions make the ∆E or ∆G values less positive, the reaction is still thermodynamically unfavorable (∆E = 6.181 kcal/mol and ∆G = 5.351 kcal/mol). The solvation energy of HCN and HSCN is contributing only ~4 kcal/mol to the ∆G (solvation of HSCN is greater). CH3S- has solvation energy only ~5 kcal/mol more than that of CH3SS-. Both of these are not sufficient to overcome the gas phase endothermicity. (B) (II) S-transfer from CH3SSH to (i) Cyanide: When sulfur atom is transferred from methyl perthiol to cyanide anion it will generate methyl thiol and thiocyanate: − − CH3SSH + CN → CH3SH + SCN . This reaction is exothermic in the gas phase (∆E = -50.42 kcal/mol and ∆G = -64 kcal/mol). The major factor behind driving this reaction is the formation of much stronger C-S bond and breaking of the weak S-S bond. Also, the gas phase PA values indicate that formation of methyl thiol (-29.64 kcal/mol) from methyl perthiol (-21.63 kcal/mol) is contributing a minor amount to 1460 INDIAN J CHEM, SEC A, SEPT-OCT 2011 the total energy. In an aqueous environment the reaction becomes energetically less feasible (from ∆G = −29.795 kcal/mol to ∆G = −43.43 kcal/mol). This +20 kcal/mol difference between the gas phase and the solvent corrected ∆G can be accounted for by the lower solvation energy of SCN- relative to the solvation energy of CN−. (ii) Hydrocyanic acid: Sulfur transfer from methyl perthiol to hydrocyanic acid will produce methyl thiol and hydrothiocyanic acid: CH3SSH + HCN → CH3SH + HSCN. This reaction is calculated to be almost thermoneutral (∆E = 1.867 kcal/mol and ∆G = 0.811 kcal/mol). Upon solvation, the reaction becomes slightly more favorable (∆E = −1.63 kcal/mol and ∆G = −2.69 kcal/mol). Slightly greater hydration energy of the products than the reactants may account for this observation. − (B) (III) S and H+ transfer from CH3SSH to – CN In addition to transfer of S atom from S donors, synchronous transfer of both S and H+ group has also been considered. When S and H+ groups are transferred from methyl perthiol to cyanide, methyl thiolate and hydrothiocyanic acid are the expected products: CH3SSH + CN− → CH3S− + HSCN. The calculations indicate that this reaction is thermodynamically feasible. (∆E = −6.195 kcal/mol and ∆G = −6.877 kcal/mol). SCN− has a lower PA (1.47 kcal/mol) than that of CH3SS− (−21.83 kcal/mol) which disfavors the reaction. However, breaking of weaker S-S bond in CH3SSH and formation of a strong C-S bond in HSCN drives the reaction. Upon inclusion of solvation correction, the reaction is calculated to be thermoneutral (∆E = 1.327 kcal/mol and ∆G = 0.654 kcal/mol.). The hydration energy of cyanide (−79.25 kcal/mol), the reactant, is much larger compared to the hydration energy of CH3S- (−65.23 kcal/mol), the product (the hydration energies of the neutral species are rather small and have minimal effect on the ∆G of the reaction). This makes the reaction unfavorable in the aqueous medium relative to the gas phase by ~7 kcal/mol. (B) (IV) S-transfer from CH3SS- and H+ transfer from HCN It is also possible that the reaction between CH3SS− to HCN yields CH3SH and SCN−: CH3SS− + HCN → CH3SH + SCN−. This reaction is favorable in gas phase (∆E = −27.606 kcal/mol and ∆G = −27.279 kcal/mol). The formation of a strong C−S bond in SCN- in place of a weaker S−S bond in CH3SS− provides the driving force for this reaction. In water, the reaction is also favorable (∆E = −24.938 kcal/mol and ∆G = −24.611 kcal/mol). The calculations presented herein suggest that biologically important sulfur transfer from thiosulfate to cyanide is thermodynamically favorable, while sulfur transfer from thiosulfate to hydrocyanic acid is unfavorable. The former is due to the formation of a strong C−S bond in SCN− (product) and the large solvation energy of SO32− (product) while the latter is mainly due to the high proton affinity of CN− (reactant) relative to SCN− (product). This presents a dilemma because in physiological pH (7.4), HCN will be present and not CN- as HCN has a pKa of 9.2. In fact, S−transfer from thiosulfate to CN− could only be achieved at pH > 10 in vitro.32 Thus, it is evident that catalysis is required to drive this process under physiological conditions. Rhodanese stores the sulfur in a cysteine residue by converting the catalytic cysteine to cysteine perthiol. The calculations indicate that this process also requires the cysteine thiol to be deprotonated. However, the pKa of thiol is ~10. Thus, in biologically relevant pHs this residue will be protonated. In the rhodanese active site, two basic residues, namely Arg 116 and Arg110, are present in the active site and they are H-bonded to the catalytic cysteine to deprotonate the cysteine thiol to make cysteine thiolate. The active site resulting after the S-transfer from thiosulfate will have a perthiolate H-bonded to a protonated arginine residue (Scheme 1, top). From here, S-transfer can occur via three pathways: (i) to CN−, (ii) to HCN, and, (iii) to HCN with a proton transfer to the resultant thiolate. Thermodynamic considerations disfavors S-transfer to HCN as it is an increase of 5.3 kcal/mol. Further, the population of CN- in pH = 7.4 buffer is < 1 %. This eliminates path (i). Thus, a synchronous S-transfer from perthiolate and proton transfer from HCN seems to be the thermodynamically favored route. However it must be mentioned that if a 2nd basic site is available to deprotonate HCN to CN- then the S−transfer can take place following path (i). Alternatively, the active site resulting from S-transfer from thiosulfate may have a perthiol H-bonded to a neutral arginine residue. Here also, there are several possible S-transfer routes (Scheme 1, bottom). However, our calculations indicate that the PA of CH3SS− is ~8 kcal/mol lower than that DEY & DEY: DFT CALCULATIONS ON BIOLOGICAL S-TRANSFER REACTIONS 1461 The possible routes for S-transfer from CysSSH to CN- in the 2nd step of the reaction. The favorable steps are indicated by √ while the disfavored ones are indicated by . ∆Gg and ∆Gs represents free energy for the process in the gas phase and after solvation correction, respectively × Scheme 1 Proposed mechanism for S-transfer for perthiol to CN- in the active site of bovine rhodanese Scheme 2 1462 INDIAN J CHEM, SEC A, SEPT-OCT 2011 of CH3S−. This implies that the pKa of CH3SS− will be 6 log units lower than that of CH3S−. In fact, cysteine perthiol is estimated to have a pKa of 6.6 as against 8.3 in cysteine. Thus, with a lower pKa than cysteine it is unlikely that cysteine perthiol will stay protonated in the presence of the same basic residue that deprotonates cysteine (cysteine has to be deprotonated to thiolate for S-transfer from thiosulfate). Thus, the possible S-transfer pathways involving a cysteine perthiol are likely to be physiologically irrelevant. Based on the thermodynamic parameters calculated for the probable pathways for S-transfer from a perthiol/perthiolate to HCN/CN− and the relative pKas of the species involved, the following mechanism (Scheme 2) can be proposed.33 An active site basic residue (either of the two conserved arginine residues H-bonded to the catalytic Cys 254 thiol sulfur of bovine rhodanese) deprotonates the cysteine thiol to thiolate, that then accepts an S-atom from thiosulfate (∆G = −18.9 kcal/mol). This reaction is driven by the large solvation energy of the resultant SO32− anion. The cysteine perthiol formed remains deprotonated as it has a lower pKa. A concerted H+ and S transfer takes place between HCN and the cysteine perthiolate. To facilitate this thermodynamically favored S-atom transfer with synchronous proton transfer, the active site needs to shuffle protons from HCN to the resultant thiol. This can be achieved utilizing the two arginine residues present where one can abstract the proton from HCN, while the other simultaneously transfers a proton to the thiolate formed after S-transfer. Thus, the two acidic arginine residues that are H-bonded to the cysteine sulfur atom plays a major role in (a) keeping the active site thiol deprotonated, and, (b) form a network of H-bonding which may help proton shuffling in the active site. References 1 2 3 4 Lang K, Biochem Z, 259 (1933) 243. Mueller E G, Nat Chem Biol, 2 (2006) 185. Brune D C, Biochim Biophys Acta, 975 (1989) 189. Wood P M, Chemolithotrophy, in Bacterial Energy, Transduction, edited by C Anthony, (Academic Press, London, UK) 1988, pp. 183-230. 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 Cipollone R, Ascenzi P, Tomao P, Imperi F & Visca P, J Mol Microbiol Biotechnol, 15 (2008) 199. Cahn R S & Dermer O C, Introduction to Chemical Nomenclature, 5th Edn, (Butterworth, London and Boston) 1979, p. 124. HeltonM E, Chen P, Paul P P, Tyeklár Z, Sommer R D, Zakharov L N, Rheingold A L, Solomon E I & Karlin K D, J Am Chem Soc, 125 (2003) 1160. Fujisawa K, Moro-oka Y & Kitajima N, J Chem Soc, Chem Commun, (1994) 623. York J T, Brown E C & Tolman W B, Angew Chem, Int Ed, 44 (2005) 7745. Bordo D & Bork P, EMBO Report, 3 (2002) 741. Bonomi F, Pagani S, Cerletti P & Cannella C, Eur J Biochem, 72 (1977) 17. Abdolrasulnia R & Wood J L, Biochim Biophys Acta, 567 (1979) 135. Ploegman J H, Drent G, Kalk K H, Hol W G J, Heinrikson R L, Keim P, Weng L & Russel J, Nature, 273 (1978) 124. Bordo D, Deriu D, Colnaghi R, Carpen A, Pagni A & Bolognesi M, J Mol Biol, 298 (2000) 691. Lu W-P, Swoboda B E P & Kelly D P, Biochim Biophys Acta, 828 (1985) 116. Friedrich C G, Rother D, Bordischewsky F, Quentmieier A & Fisher, J. Appl Environ Microbiol, 67 (2001) 2873. Quentmeier A & Friedrich C G, FEBS Lett, 503 (2001) 168. Cipollone R, Ascenzi P & Visca P, IUBMB Life, 59 (2007) 51. Horowitz P & Criscimagna N L, J Biol Chem, 258 (1994) 7894. Pagani S, Forlani F, Carrpen A, Boedo D & Colnaghi R, FEBS Lett, 472 (2000) 307. Cipollone R, Bigotti M G, Frangipani E, Ascenzi P & Visca P, Biochem Biophys Res Comm, 325 (2004) 85. Aminlari M & Gilanpour H, Comp Biochem Physiol, B99 (1991) 673. Pagani S, Bonemi F & Cerletti P, Eur J Biochem, 142 (1984) 361. Perdew J P, Phys Rev B, 33 (1986) 8822. Becke A D, J Chem Phys, 84 (1986) 4524. Perdew J P, Burke K & Ernzerhof M, Phys Rev Lett, 77 (1996) 3865. Becke A D, J Chem Phys, 98 (1993) 5648. Lee C, Yang W & Parr R G, Phys Rev B, 37 (1988) 785. Miertus S, Scrocco E & Tomasi, J Chem Phys, 55 (1981) 117. Bjerrum J, Stability Constants, (Chemical Society, London) 1958. Muchenberg U, Anwar A, Mechlenberg S & Jacob C, Org Biomol Chem, 5 (2007) 1505. Westley J & Heyse D, J Biol Chem, 246 (1971) 1468. Note that these H-bonding interactions are not modeled in the current study. These are very important and will be included in future calculations.