Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
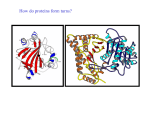
From www.bloodjournal.org by guest on June 17, 2017. For personal use only. The Role of Amino-Terminal Residues of the Heavy Chain of Factor IXa in the Binding of Its Cofactor, Factor VIIIa By Nobuko Hamaguchi, S. Paul Bajaj, Kenneth J. Smith, and Darrel W. Stafford The purpose ofthis study isto determine which residues of the factor IXa heavy chain areimportant for interactionwith the cofactor of factor IXa, factor Vllla. Because the monoclonal antibody(MoAb) FXCOO8 inhibitsinteractionbetween factors IXa and Vllla, and becauseit also reactswith residues 181-310 ofthe factor IXa heavy chain,we usedthe computermodelled structure of the factor IXa heavy chain to select charged surface residues likely to interact with FXCOOZ)and/ or factor Vllla.We made mutations in the region of residues 181-310 of the heavy chain of factor IX, and replaced these amino acids individuallywith those located at the same position in factor X. The mutated factor IX retained complete clotting activity and thus interacted normally with factor Vllla. Five mutant proteins (factorIXW,W,factor IX-, factor IXE2400, factor lXIUIIV, and factor IX-1 reacted with heavy chain-specificMoAbr FXCOO8 and A-5.Neither factor l&2,M norfactorIXWm bound to FXCOO8. Factor had reduced affinity to FXCOOZ). Our results suggestthe following: (1) factor IXa residues 214,228,240,247,248,252,260, and 276 are not involved in specific interaction with factor Vllla; and (2) the FXCOO8 and factor Vllla binding sitesmay not share critical residues. 0 1994 by The American Society of Hematology. F recombinant techniques, we made FIXs with various amino acid substitutions in the heavy chain region. Because intermolecular interactions commonly involve solvent-accessible surface regions, the water-accessible surface area of each residue of FIX was calculated from the computer model of the catalytic domain of FIXa based on trypsin x-raystructure. These calculations were used to select candidates for the critical residues for FXC008binding. Even though a large part of the intermolecular binding energy is from hydrophobic interactions, the complementarity of hydrophobic interactions is relatively inducible, and the specificity of intermolecular interaction is mainly achieved by hydrogen bondelectrostatic interactions. Thus to decrease the affinity of two proteins for each other, it is reasonable to alter the electrostatic properties of the surface residues. Based on the structural model of the heavy chain of FIXa, seven charged surface residues between amino acids 181 and 310 were selected for creating mutant proteins. In addition, residue 260 was mutated because this residue is located on the surface, and a naturally occurring mutation at this position is known to cause hemophilia B.’ To minimize structural distortion in the mutant molecules, FIX residues were individually replaced by FX residues from the homologous position. The mutant molecules were characterized by MoAb binding and characterized for functional properties. ACTOR (F)IX is the precursor of the protease FIXa required for blood coagulation. People with defective FIX molecules suffer from hemophilia B, an X-linked hereditary bleeding disorder.’ The structural organization of FIX is similar to the other vitamin K-dependent blood coagulation proteins including FX and FVII. The light chain of FIXa contains the N-terminal 145 amino acids of FIX. It consists of the y-carboxyglutamic acid (Gla) domain, necessary for binding to negatively charged phospholipid membranes and to endothelial cells,’ the hydrophobic domain, and the two epidermal growth factor-like (EGF) domains. The EGF domains have been suggested to be parts of the FVIIIa binding site.3 Sequence analysis of several species has shown that the activation peptide (residues 146-180), which is released on conversion of FIX to FIXa, is the least conserved region among the vitamin K-dependent zymogens. The heavy chain (residues 181-415) of FIXa contains the active site triad of histidine 221, aspartic acid 269, and serine 365. The amino acid sequence of the catalytic domain of FIXa is highly homologous to other serine proteases such as trypsin, chymotrypsin, and thrombin. Approximately 70% of the point mutations in FIX that cause hemophilia B are found in the heavy chain region of the m01ecule.~Some mutations directly affect FIX function, whereas others may affect the structural stability of the molecule. Possible mechanisms of the functional defects can be divided into three classes: (1) defects in the proteolytic activity; (2) defects in the substrate (FX) and cofactor (calcium, FVIIIa, and platelets) recognition sites; and (3) defects in the activation by FXIa and the FVIIa-tissue factor complex. The anti-FIX monoclonal antibody (MoAb) FXC008 has been reported to interfere with the binding of FIXa to F V I Ea.’ FXC008 binds to an immunodominant site in the heavy chain of FIXa between residues 181 and 310.6 These results led to the suggestion that the FVIIIa binding site may reside between residues 181 and 310 of FIXa. Because the affinity of FIXa for FVIIIa in the absence of a surface membrane is known to be weak (kd of 1 pmol/L), and because activated FVIIIa is very unstable, it has been difficult to measure the binding between these two macromolecules. Thus, we chose an alternative approach to investigate FIXalFVIIIa interactions by further defining the epitope of FXC008 in the heavy chain and determining if the same residues are involved in FVIIIa recognition by FIXa. To accomplish this using - Blood, Vol 84, No 6 (September 15). 1994: pp 1837-1842 From the Department of Biochemistry, Duke University Medical Center, Durham, NC; the Departments of Medicine and Pathology, University of New Mexico School of Medicine and United Blood Services, Albuquerque, NM; the Department of Biochemistry, St Louis University, St Louis, MO; and the Department of Biology and Center for Thrombosis and Hemostasis, University of North Carolina at Chapel Hill, NC. Submitted January 19, 1994; accepted May 6, 1994. Supported by National Institutes of Health Grant No. R01 HL38973, and by a grant from Blood Systems Research Foundation, Scottsdale, AZ. Address reprint requests to Darrel W. Stafford, PhD, Department of Biology and Center for Thrombosis and Hemostasis, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3280. The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact. 0 1994 by The American Society of Hematology. 0006-497I/94/84W-Oll5$3.00/0 1837 From www.bloodjournal.org by guest on June 17, 2017. For personal use only. 1838 HAMAGUCHI ET AL Table 1. Oligonucleotides Used for In Vitro Mutagenesis Position 214 228 240 247 248 252 260 276 Mutation K-F K-R E-.Q K-V R-H R-V N-T D-K Oligonucleotide (5' - 3') CGlTAATGAAlTCGTGAlTGTAAC GAAACTGGTGTACGTAlTACAGlTGTC CATAATATTGAGCAGACAGAGCACACAGAGCAMAG ACAGAGGAAGlTCGAAATGTG GAGCAAAAGCATAACGTGATTCGA CGAAATGTGAlTGTAATCATCCCTCACCACAAC CACAACTACCACAGCTGCTATTAATAAG GCCCTTCTGGAGCTCAAAGAAACCCTTAGTG based on the bovine thrombin x-ray crystallographic structure and energy-minimized using Amber version 3.1 (Department of Pharmacy, University of California, San Francisco) with explicit solvent using periodic boundary condition. The solvent-accessible surface area was calculated using the Determination of Secondary Structure of Protein (DSSP) program (Brookhaven National Laboratory,Upton, NY).I3 RESULTS AND DISCUSSION Selection of the charged sugace residues in factor 1x for the study o f factor VIIIa binding. The purpose of this study MATERIALS AND METHODS wastodeterminewhether the binding of FIX to M I I I a The oligonucleotides used for in vitro mutagenesis were purchaseddepends on the LlX-specific residues required for binding fromOligo(Wilsonville, OH) (Table 1). Theseoligonucleotides FIX to MoAb FXCOO8. The alignment of the amino acid (except for an oligonucleotide mutation at residue 247) were also designed to createnew restriction enzyme recognition sites; EcoRI, sequence of the catalytic domain of FIXa and other related serine proteases was based on published topologic similariSnaBI,HgiAl,MaeII, FokI, PvuIIorSacI. 'l7 DNApolymerase ties between residuesin thrombin and ~hymotrypsin.'~ There (Sequenase),polynucleotidekinase,andT4 DNA ligase were obare 25 charged residues between residues 181-310. These tained from Bethesda Research Laboratory (Bethesda, MD), New residues are likely to be on the surfaceof the molecule England Biolabs (Beverly, MA), and Boehringer Mannheim (Chirespectively. [12SI]Protein A, [12sI]Na, and [%]dATP were and are thereforepotentially involvedinmacromolecular cago, L), L), Afpurchased from Amersham Corporation (Arlington Heights, interactions. Based on the high homology among enzymes fiPrep 10 was obtained from Bio-Rad (Richmond, CA), Iodobeads and cofactors in coagulation factors, it is verylikely that fromPierce(Rockford,IL),andGeneticin(G418)fromGibco these factors interact in a similar manner and that a few (Grand Island, NY). FIX was purified from pooled plasma as despecific interactions between correct partners such as FIXascribed previously.8 FIX-deficient plasma was purchased from Sigma FVIIIa and F X a - M a are responsible for the recognition. At (St Louis, MO). All other reagents were of the highest purity comthe same time, these interactions are conserved among spemercially available. cies (cow, mouse) because the clotting time of human FIXThe anti-human FIX MoAbs used in this study are: A-l, A-5, A7, and FXC008. The amino acid sequences of human FIX recognized deficient plasma can becorrected by FIX from otherspecies. by these antibodies are:A-l in residues 147-153 (activation peptide), As candidates of residues involved in FIXa-specific interacand A-5 and FXC00S in residues 181-310 (the heavy chain). A-7 is tion with FVIIIa, the following charged residues were omita calcium-dependent conformation specific antibody that recognizes ted in this study because they are either identical in FIX amino acids 1-42 (the Gla domain). and FX or not conserved among species: aspartic acid 186 In vitromutagenesis and construction of expressionplasmids. (asparagine in mouse), lysine 188 (glutamic acid in cow), Site-directed mutagenesis was performed as described by Kunkel.' mouse), glutamicacid 213, lysine 201 (glutamicacidin Each mutated cDNA was sequenced by the dideoxy-chain terminaglutamic acid 239 (aspartic acid in mouse), glutamic acid tion method," and mutated cDNA was inserted into pCMV4 vector." 242, and aspartic acid 269. Production of recombinant FIX. Humanembryokidney293 The computer modelof the catalytic domainof FIXa was cells were grown in a mixture of Dulbecco's modified Eagle's meused topredictthe water-accessible surfacearea of the diumand F-l2 medium (1:l) supplementedwith10%fetalcalf ~ r 0 g r a m . I(Fig ~ 1). The serum. pCMV4 FIX expression vectors were co-transfected into cellscharged residues using the DSSP with pSV2neo (selection marker plasmid) using the calcium-phosresults indicate that residues235 and 274, which are respecphate co-precipitation method. Selectionof cells for production and tively equivalent to 70 and 107 in p p s i n , have low waterpurificationofrecombinant FIxs fromculturemediumwaspreaccessible surface areas (16 and 35 A', respectively). This reviously described.12Stable cell lines expressing each of the different sult is consistent with thex-ray crystallographic structure of recombinant FIX moleculesproducedseveralhundredpg/L/din trypsin (Brookhaven Protein Data Base, PDB2TPT, 11 and roller bottles. Recombinant FIXs were purified from culture media 36 A' for residues 70 and 107, respectively). Thus the data throughanA-7MoAbcolumn,concentratedusing Centriprep 30 on water accessibility suggest that residues 235 and 274 were (Amicon,Danvers,MA),anddialyzedagainstTBS (10 m m o E not directly involved in binding to other macromolecules. Tris, 150 m m o E NaCl, pH 7.2) overnight. The epitopeof FXCOO8, reported to inhibitM I I I a binding of Iodination of protein and radioimmunoassay.Iodination to FIXa, requires at least 130 residues for mapping.6 This MoAb and radioimmunoassays (RIA) performed by the sandwich method have been described previously.12 Either A-5,A-l, or FXCsuggests thatthe epitope for this MoAb is discontinuous and/ 008 MoAb was used to coat the wells and 1251-labeled A-7 MoAb or requiresspecific internal residues to support the structure. was used to detect the bound FIX antigen level in each well. Based on this assumption, we eliminated residues185,224, Clottingassay.One-stageactivatedpartialthromboplastintime 265, and 301 as primary candidates for mutation because (m) assays were performed according to the manufacturer's inthese residues are located at the edge of the surface constructions (Organon Teknika, Durham, NC). The abilityof the protaining the epitope (residues 181-310, shown in white in Fig teins to correct the clotting time of FIX-deficient plasma was calcu2A). lated from a standard curve derived from pooled normal plasma. There are a number of point mutations in the catalytic Molecular modeling. The original model for factor IX was dedomain of FIX that cause hemophilia B. The majority of veloped from the x-ray crystallographic coordinatesof bovine panknown mutations are internal, nonpolar residues such asalacreatic trypsin as described previously." The structure was modified From www.bloodjournal.org by guest on June 17, 2017. For personal use only. IX HEAW CHAIN IMMUNODOMINANT SITES 200 1839 I I too 0 residue number Water-accsdble s u h c e area of s e l e c t e d residua in the catalytic domain of FIXa. The water-accsdble surface orear r a i d u r within fint 130 residua of tho catrrlytk domain calculated uringDSSP." nine 220 to valine," and alanine 233 to threonine,I6 and seem likely to destabilize the molecule. However, some defective FIXs seem to have a point mutation on the surface; for example, asparagine to serine at 260 and threonine to methionine at 296. The antigen level of FIX with a point mutation at residue 296 is reported to be less than 10%of normal. Thus, mutations at this position probably affect the overall stability of the molecule. The plasma antigen level of defective FIX with a mutation at 260 has not been reported7; it was therefore included in this study. Finally, based on the above considerations, residues at 214,228,240, 247,248,252,260,and 276 were exchanged for residues found at the equivalent positions in FX (shown in purple in Fig 2A). Each mutation in the FIX cDNA was accomplished by in vitro mutagenesis using the oligonucleotides listed in Table 1. Clotting activity and reactivity with MoAb of mutatedfactor Ixs. The results of the clotting assay and RIA are shown in Table 2. F I X D 2 7 6 K bound 15% as well as plasma FIX to MoAb A-5. The same assay using FXC008 failed to detect binding Of m D ' 2 7 6 K and F I X R X ~ ~F .I X ~ 2 5 2 vhad reduced affinity for FXC008.Our results with F I X R X s H are similar to those obtained with the m m 4 8 9 mutant with respect to MoAb FXC008 binding.I7 Overall, the results indicate that the epitopes of A-5 and FXCOO8 overlap, but are not identical. In the computer model, residues 248, 252, and 276 are linearly arranged with residue 276 in the center. The distances between alpha carbonsoof residue 276 and residues 248 and 252 are 6.4, and 8.6 A, respectively (Fig 2B). Be- of charaed - cause these residues are located in the center of the surface defined by residues 181-310 of the catalytic domain, they must be part of the epitopes for several known MoAbs such as A-4, A-5, CD10, and FXC008.6 However, in the one stage ap?T assay, mmgH, FIXRzszv, and m D 2 7 6 K retained normal activity. After preincubation had prowith excess FXC008, normal FIX and longed clotting time, whereas the clotting times of F I X m H and were unaffected by the presence of FXC008. This result is consistent with the results from FUA. If any of the charged residues that were changed made contact with FVIIIa, then a change of at least 3 kcal per mole in binding energy would be expected." The concentration of M I I I in plasma is estimated to be about 0.5 nmoyL. This concentration is close to the dissociation constant estimated for IXa-VIIIa in the presence of phospholipid membrane^.'^ It was shown with a chimera molecule (m) that a slightly reduced (15-fold) affinity to M a of this chimera resulted in significantly reduced FXa physiologic activity in the presence of membrane.20 Therefore the changes that affect the FIXa specific interaction with MIIIa should dramatically affect the FIXa procoagulant activity. Because no activity was lost, we can conclude that these residues have no critical direct role in binding FVIIIa. The interaction of an antibody with a prottin antigen might involve surface areas witha25 to 30 A diameter containing 13 to 16 residues on each molecule." Accordingly, the binding site for FXC008 must interface with residues other than these three residues. Mutated residues 214 From www.bloodjournal.org by guest on June 17, 2017. For personal use only. HAMAGUCHI ET AL 1840 L- - Fig 2. FlXa computer model showing residues relevant to this study. (A) The relative positions of the active site, the binding site for EGF2 (based on the FXa structure)," and the surface residues constituting part of the FXCOO8 MoAb binding site. The backbone ribbon ofresidues 181-310 is shownin blue, and thatof residues 311415 in green. Residues, including side chains, of the catalytic triad are shown in red. Side chains of residues likely t o contact the EGF2like domain are depicted in green at the bottomof the figure. The side chains of residues subjected t o in vitro mutagenesis are shown in purple. Residues that differ between FIX and FX, but that were omitted from this studybecause of their positions, are depicted in white. (B) A side view of Fig 2A. The backbone ribbon of residues 181-310 is blue, and thatof residues 311-415 is yellow. Mutatedresidues are shown in purple. Residues in the catalytic triadare in red. A calcium ion in the putativecalcium binding loop is shown in green between glutamic acid residues 235 and 245 (in yellow). and 228 are located close to resjdue 276 with inter-alpha carbon distances of 7.7 and 11.7 A, respectively. These residues are linearly arranged with residue 276 in the center, and perpendicular to residues 248 and 252 (Fig 2B). Mutations at 214 and 228 had no effect on clotting activity or binding to FXC008. These results imply that charged residues 248,252, and 276 play a key role in binding to MoAb FXC008 and that the epitope consists of discontinuous sequences. Bajaj et al showed that a synthetic peptide corresponding to FIX residues 231-265 binds MoAb FXC008 with 5Clo-fold reduced affinity." The reduced affinity may be caused by the peptide missing a part of the epitope containing residue 276 and adjacent residues. Wildgoose et al reported that a peptide containing the sequence corresponding to residues 195-206 of FVII inhibits the interaction between FVII and tissue factor.z3This region is equivalent to residues 223-231 in FIX and is close to histidine in the catalytic triad of FVII and FIX. In the x-ray crystallographic structure of thrombin, this region is found to have a 10-amino acid insertion compared with chymotrypsin and forms a loop structure protruding into the active site cleft. This presumably affects the substrate and inhibitor ~pecificity.'~ If this region of FIX were responsible for substrate specificity, it would presumably not be involved in the FVIIIa binding site because it would compete with the substrate. The mutation in the center of this region in FIX, lysine to arginine at position 228, did not affect its clotting activity. Furthermore, the sequence of residues 223-228 of FIX is not conserved among different ~pecies.2~ These data imply that residues 223-23 1 may not be directly involved in FIX specific functions. and F I X N z m had reduced affinity for the MoAb A-l, whose epitope was mapped to residues 147-153 of the activation peptide: These mutants had approximately 50% and 80% affinity for A-l, respectively, compared with A-5. This suggests that residues 214 and 260 may be located topologically close to the activation peptide. It would be interesting to know how the activation peptide connects the light chain and the heavy chain. This would aid in understanding the domain construction of FIX, the structural importance of the activation peptide, and the activation process. Among the residues changed from FIX to FX in this study, residues 248, 252, and 260 are found at positions known to cause hemophilia B. Several hemophilia B patients have arginine 248 substituted by glutamine at residue 248.15.17.z5 In our study, arginine 248 was changed to histidine, the Table 2. Clotting Activity and Antigenicity of FlXs With Mutations in an lmmunodominant S i e in the Catalytic Domain FXCOOB and and A-l Mutants K214F K228R E240Q K247V R248H R252V N260T D276K A-7* (%) 52 2 2 95 ? 10 902 3 90 ? 6 89 2 4 94 t 1 79 2 3 92 ? 5 A-5 and A-7t (%) 97 ? 1 95 ? 5 98 ? 2 91 ? 2 93 ? 1 95 2 1 90 2 8 13 ? 3 a P T Assay (%) 1%) -100 -100 -100 90 2 4 85 2 5 94 ? 5 90 5 8 86 2 4 94 ? 5 95 ? 3 92 2 4 -100 ND -20 -100 ND Antigenicities are shown as relative to plasma-purified FIX; normal in aPlTassay indicates 80% to 100% of plasma-purified FIX. Abbreviation: ND, not detected. The wells were coated with A-l MoAb and '251-labeled A-7MoAb was used as a second antibody. t The wells were coated with A-5 MoAb and '251-labeled A-7MoAb was used as a second antibody. +The wells were coated with FXCOO8 MoAb and '251-labeled A-7 MoAb was used as a second antibody. The data were obtained from a single experiment; however, similar results were obtained from Western blotting with FXCOO8as an antibody. From www.bloodjournal.org by guest on June 17, 2017. For personal use only. FACTOR IX HEAW CHAIN IMMUNODOMINANT SITES residue found in FX at the equivalent position. This change had no effect on the procoagulant activity, indicating that this residue has no specific Fu( function. This observation agrees with that of Ludwig et al, who found that purified FIX from a patient with glutamine at 248 had 41% normal activity.I7 The mutation at 252 found in a patient was arginine to leucine: whereas the substitution in our study is valine. Another mutation at 260 found in patients was asparagine to serine7; the substitution in the present study is to threonine. The antigen level of FIX in patients with mutations at 252 or 260 has not been reported. Because our results indicate that these residues are not critical for the clotting activity, these naturally occumng mutations may cause the defect by affecting the half-life in circulation of the molecule. Alternatively, the known mutations may cause a global conformational change rendering its physiologic function inactive. It has been shown that platelet binding of a FIX mutant with its second EGF domain changed to that of FX, is enhanced by FVIIIa.26This indicates that the FIX molecule with the second EGF domain from FX still recognizes FVIIIa. Thus, the second EGF domain may not be solely responsible for the binding to FVIIIa. This leads to the previously proposed hypothesis that the heavy chain and the light chain share the FVIIIa binding site.3 Inthis case, it is possible that the immunodominant site of the catalytic domain of FIXa contributes to the binding of FVIIIa, but that the residues in FX can be substituted without changing the FVIIIa binding capability. The other possible explanation is that the binding of FXC008 has an allosteric effect on other parts of the surface that involve the FVIIIa binding. It is known that allosteric effects from binding between macromolecules can be shown as far away as 20 A.” Because the activation of FIX is required before its binding to FVIIIa, the part of the catalytic domain that becomes solvent accessible on the release of the activation peptide may play acritical role in the binding to FVIIIa. In this case, the binding of FXC008 may be disrupting the normal conformational changes that occur in other parts of the heavy chain surface after activation. Three residues (R248, R252, D276) that are found to be responsible for FXC008 binding are also found in mouse FIX.27Nonetheless, mouse FIX does not react with MoAb FXC008. This means that there are other crucial structural determinants in human FIX that play a substantial role in binding to the MoAb. The additional determinants for MoAb FXC008 may be shared in part by the FVIIIa binding region. It will be necessary to take several different approaches to thoroughly understand the interaction between FIXa and FVIIIa. ACKNOWLEDGMENT We would like to thank Dr Paul Charifson for help in molecular modeling and Dr David Straight and Byron Woods for reviewing the manuscript. REFERENCES 1. Mann KG, Neshiem ME, Church WR, Haley P, Krishnaswamy S: Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76:1, 1990 1841 2. Cheung W, Hamaguchi N. Smith KJ, Stafford DW: The binding of human factor M to endothelial cells is mediated by residues 3-11. J Biol Chem 267:20529, 1992 3. Astermark A, Stenflo J: The epidermal growth factor-like domains of factor IX. J Biol Chem 266:2438, 1991 4. Ginnelli F, Green PM, High KA, Sommer S, Lillicrap DP, Ludwig M, Olek K, Reitsma PH, Goossens M, Yoshioka A, Brownlee GG: Haemophilia B: Database of point mutations and short additions and deletions, ed 4, 1993. Nucleic Acids Res 21:3075, 1993 5. Bajaj P, Rapaport SI, Maki SL: A monoclonal antibody to factor M that inhibits the factor VII1:Ca potentation of factor X activation. J Biol Chem 26011574, 1985 6. Frazier D, Smith K, Cheung W, Ware J, Lin S, Thompson A, Reisner H, Bajaj P, Stafford D: Mapping of monoclonal antibodies to human factor M.Blood 74:971, 1989 7. Koeberl DD, Bottema CDK, Buerstedde JM, Sommer SS: Functionally important regions of the factor IX gene have a low rate of polymorphism and a high rate of mutations in the dinucleotide CpG. Am J Hum Genet 45448, 1989 8. Smith KJ, Ono K: Monoclonal antibodies to factor IX: Characterization and use in immunoassays for factor IX. Thromb Res 33:211, 1984 9. Kunkel TA: Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA 82:488, 1985 10. Tabor S , Richardson CC: DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc Natl Acad Sci USA 84:4767, 1987 11. Anderson S, Davis DL, Dahlback H, Jornvall H, Russell DW: Cloning, structure and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem 264:8222, 1989 12. Hamaguchi N, Charifson PS, Pedersen LG, Brayer GD, Smith JK, Stafford DW: Expression and characterization of human factor IX. J Biol Chem 266:15213, 1991 13. Kabsch W, Sander C: Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577, 1983 14. Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J: The refined 1.9 A crystal structure of human a-thrombin: Interaction with D-Phe-Pro-Arg chloromethyl ketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J 8:3467, 1989 15. Koeberl DD, Bottema CDK, Ketterling RP, Bridge PJ, Lillicrap DP, Sommer S S : Mutations causing hemophilia B: Direct estimate of the underlying rates of spontaneous germ-line transitions, transversions and deletions in a human gene. AmJHum Genet 47:202, 1990 16. Green PM, Montandon AJ, Ljung R, Bentley DR, Kling SR, Nilsson IM, Giannelli F: Hemophilia B mutations in a complete Swedish population sample: A test of new strategy for the genetic counselling of diseases with mutational heterogeneity. Br J Haematol 78:390, 1991 17. Ludwig M, Sabhanval A, Brackmann HH, Olek K, Smith KJ, Birktoft JJ, Bajaj S P Hemophilia B caused by five different nondeletion mutations in the protease domain of factor M.Blood 79:1225, 1992 18. Fersht AR, Shi J, Knill-Jones J, Lowe DM, Wilkinson M , Blow DM, Brick P, Carter P, Wayne MMY, Winter G: Hydrogen bonding and biological specificity analysed by protein engineering. Nature 314:235, 1985 19. Kane WH, Davie EW: Blood coagulation factors V and VIII: Structural and functional similarities and their relationship to hemorrhagic and thrombotic disorders. Blood 71539, 1988 20. Herzberg MS, Bel-Tal 0, Furie B, Furie B: Construction, From www.bloodjournal.org by guest on June 17, 2017. For personal use only. 1842 expression, and characterization of a chimera of factor IX and factor X. J Biol Chem 267:14759, 1992 21. Colman PS: Structure of antibody-antigen complexes: Implications for immune recognition. Adv Immunol 43:99, 1988 22. Bajaj SP, Sabharwal AK, Gorka .I, Birktoft A: Antibodyprobed conformational transitions in the protease domain of human factor IX upon calcium binding and zymogen activation: Putative high-affinity Caz+-binding site inthe protease domain. Proc Natl Acad Sci USA 89:152, 1992 23. Wildgoose P, Kazim AL, Kisiel W: The importance of residues 195-206 of human blood clotting factorVII in the interactionof factor W with tissue factor. Proc Natl Acad Sci USA 87:7290, 1990 24. Sarkar G, Koeberl DD, Sommer SS: Direct sequencing of the activation peptide and the catalytic domain of the factor IX gene in six species. Genomics 6:133, 1990 HAMAGUCHI ET AL 25. Chen S-H, Zhang M, Lovrien EW, Scott CR, Thompson AR: CG dinucleotide transitions in the factor IX gene account for about half of thepoint mutations in hemophilia B patients: A Seattle series. Hum Genet 87:177, 1991 26. Ahmad SS, Rawala-Sheikh R, Cheung W, Stafford DW, Walsh PN: The role of growth factor domains of human factor IXa in binding to platelets and in factor X activation. Thromb Haemost 65300, 1991 27. Wu S-M, Stafford DW, Ware J: Deduced amino acid sequence of mouse blood coagulation factor IX. Gene 86:275, 1990 28. Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Heiber R, Blankenship DT, Cardin AD, Kisiel W: Structure ofhuman Des(1-45) factor Xa in 2.2 A resolution. J Mol Biol 232:947, 1993 From www.bloodjournal.org by guest on June 17, 2017. For personal use only. 1994 84: 1837-1842 The role of amino-terminal residues of the heavy chain of factor IXa in the binding of its cofactor, factor VIIIa N Hamaguchi, SP Bajaj, KJ Smith and DW Stafford Updated information and services can be found at: http://www.bloodjournal.org/content/84/6/1837.full.html Articles on similar topics can be found in the following Blood collections Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.