Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
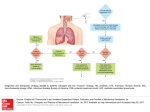
VAP: A VERSATILE AGGREGATE PROFILER FOR EFFICIENT GENOMEWIDE DATA REPRESENTATION AND DISCOVERY COULOMBE C, POITRAS C, NORDELL-MARKOVITS A, BRUNELLE M, LAVOIE MA, ROBERT F, JACQUES PÉ. (2014) Christian Poitras IRCM WHAT IS CHIP-SEQUENCING ¢ Combination of chromatin immunoprecipitation (ChIP) with ultra high-throughput massively parallel sequencing ¢ Allows mapping of protein-DNA interactions in vivo on genome scale CHIP-SEQ * Jkwchui - Cell diagram adapted from LadyOfHats' Animal Cell diagram. Information based on Illumina data sheet, as well as ChIP and immunoprecipitation articles & references. WHAT IS VAP ¢ Stands for “versatile aggregate profiler” ¢ Generates aggregate profiles of genomic datasets over groups of regions of interest ¢ Uses absolute or relative method ¢ Customizable number of windows over a specified number of reference points Reference points delimit the genes of interest as well as their flanking genes, or even exons ¢ Accessible through both a user-friendly platformindependent Java interface or via command line ¢ 3 modes: Annotations, Exons and Coordinates ¢ Can run on laptops CHIP-SEQ ANALYSIS WORKFLOW VAP VAP WORKFLOW Input • BedGraph, WIG, BigWig • Annotations (optional) • Annotation groups • Filters (optional) VAP • Version 1.1.0 (2015) Data does not need to be ChIP-Seq or ChIP-chip although this is the most common use Output • Aggregate profiles (Text, PNG) • Heatmaps (Text, PNG) ANNOTATION GROUPS ¢ For annotation and exon mode, the annotation groups files contain gene names present in the annotations file ¢ For coordinates mode, the annotation groups files contain coordinates for 2 reference points For more than 2 reference points, a special format is required ¢ VAP can create aggregate profiles of any data that have coordinates WHAT IS ABSOLUTE METHOD AND WHY SHOULD I CARE? ¢ Relative method: Windows are larger for long genes All genes are divided into the same number of windows ¢ Consequently, a signal appearing at the same distance from the TSS (e.g. 200 bp) for a long and a short gene will be placed in different windows (e.g. first window of a 4 kb gene compared to the 20th window of a 400 bp gene divided into 40 windows) ¢ Easier to program ¢ Absolute method: Windows are the same size for all genes Some windows will not exist for small genes RELATIVE VS ABSOLUTE * Pokholok D, Harbison C, Levine S, Zeitlinger J, Lewitter F, Gifford DK, Young RA. (2005) RELATIVE VS ABSOLUTE ¢ Erroneous conclusion “Long genes depend on the Set2/Rpd3S pathway for accurate transcription” due to relative method * Li B, Gogol M, Carey M, Pattenden SG, Seidel C, Workman JL. (2007) VAP OUTPUT VAP RAW AGGREGATE PROFILE (BASED ON TEST DATA) * Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. (2005) * Guillemette B, Bataille AR, Gévry N, Adam M, Blanchette M, Robert F, Gaudreau L. (2005) VAP RAW HEATMAP (BASED ON TEST DATA - UNSORTED) * Guillemette B, Bataille AR, Gévry N, Adam M, Blanchette M, Robert F, Gaudreau L. (2005) VAP INTERFACE VAP INTERFACE VAP INTERFACE THANKS ¢ Pierre-Étienne Sherbrooke Jacques ¢ Charles Coulombe – Sherbrooke ¢ François Robert – IRCM ¢ And all other authors! ¢ Benoit Coulombe Lab - IRCM