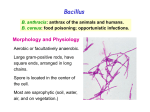
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
FEMS Microbiology Reviews 29 (2005) 303–329 www.fems-microbiology.org Genomics of the Bacillus cereus group of organisms q David A. Rasko a, Michael R. Altherr b, Cliff S. Han b, Jacques Ravel a a,* The Institute for Genomic Research, 9712 Medical Center Drive, Rockville, MD 20850, USA b Los Alamos National Laboratory, P.O. Box 1663, Los Alamos, NM 87545, USA Received 5 November 2004; received in revised form 22 December 2004; accepted 23 December 2004 First published online 28 January 2005 Abstract Members of the Bacillus cereus group of organisms include Bacillus cereus, Bacillus anthracis and Bacillus thuringiensis. Collectively, these organisms represent microbes of high economic, medical and biodefense importance. Given this significance, this group contains the highest number of closely related fully sequenced genomes, giving the unique opportunity for thorough comparative genomic analyses. Much of the disease and host specificity of members of this group can be attributed to their plasmids, which vary in size and number. Chromosomes exhibit a high level of synteny and protein similarity, with limited differences in gene content, questioning the speciation of the group members. Genomic data have spurred functional studies that combined microarrays and proteomics. Recent advances are reviewed in this article and highlight the advantages of genomic approaches to the study of this important group of bacteria. 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved. Keywords: Bacillus cereus; Bacillus anthracis; Genome; Plasmid Contents 1. 2. 3. 4. 5. 6. q * Introduction – The Bacillus cereus group of organisms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.1. Bacillus thuringiensis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.2. Bacillus cereus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.3. B. anthracis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Molecular identification of members of the B. cereus group of organisms: One species on the basis of genetic evidence? . . . Genome sequencing projects completed and in progress. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . B. anthracis genomic analysis for molecular and forensic epidemiological purposes . . . . . . . . . . . . . . . . . . . . . . . . . . Comparison of the chromosome sequences of the B. cereus group of organisms. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.1. Non-pathogenic B. cereus ATCC 14579 and B. cereus ATCC 10987 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.2. The pathogenic members of the B. cereus group of organism, B. cereus G9241, B. cereus Zebra Killer, and B. thuringiensis 97-27 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . The B. cereus group of organisms plasmid sequence comparisons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6.1. General features of the B. cereus group plasmids identified in genome sequencing. . . . . . . . . . . . . . . . . . . . . . . 6.2. Replication and mobility mechanisms of the B. cereus group plasmids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Edited by Mark J. Pallen. Corresponding author. Tel.: +1 301 795 7884; fax: +1 301 838 0208. E-mail address: [email protected] (J. Ravel). 0168-6445/$22.00 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved. doi:10.1016/j.femsre.2004.12.005 304 304 304 304 305 306 309 309 309 312 313 313 317 304 7. 8. 9. D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 The phage of the B. cereus group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7.1. Genomic phage content . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7.2. Lytic induction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7.3. Exploitation of phage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Functional genomics of the B. cereus group of organisms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8.1. Transcriptional analysis using microarrays. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8.2. Proteomics studies. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Summary: One species on the basis of genomic evidence? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1. Introduction – The Bacillus cereus group of organisms The Bacillus cereus group of organisms contains Bacillus thuringiensis, Bacillus anthracis and Bacillus cereus (sensu stricto). This group of Gram-positive sporeformers forms a highly homogeneous subdivision of the genus Bacillus. Demonstration of their high genetic relatedness has contributed to the suggestion that B. anthracis, B. cereus and B. thuringiensis are members of a single species, B. cereus sensu lato [1–3]. Traditionally, these organisms have been differentiated based on their phenotypic characteristics, including pathogenic potential. Bacillus mycoides, Bacillus pseudomycoides and Bacillus weihenstephanensis are also members of the B. cereus group of organisms. However, as no genomic data are available, these species have not been included in this review. 1.1. Bacillus thuringiensis B. thuringiensis has long been regarded as an insect pathogen alone. The insecticidal spectrum varies within the 82 different serotypes reported [4], and affects insects primarily from the orders Lepidoptera, Diptera and Coleoptera. There are also reports of B. thuringiensis isolates active against mosquitoes that are vectors for disease, such as malaria and yellow fever [5]. B. thuringiensis spore preparations have been successfully commercialized as biopesticides. The spores are associated with large crystal protein inclusions, which can make up to 25% of the dry weight of the spore preparations. The crystals are aggregates of a large protein (about 130–140 kDa) that is actually a protoxin. Upon ingestion by insect larvae, the protoxin crystals solubilize in the mid-gut, where it is cleaved by a gut protease to produce an active toxin (d-endotoxins) of about 60 kDa. The toxin binds to the mid-gut epithelial cells, creating pores in the cell membranes. As a result, the gut is rapidly immobilized and the epithelial cells lyse. The insect larva stops feeding and often dies from lethal septicemia [6]. Little is known about the ecology of B. thuringiensis and conflicting reports are reviewed by Jensen et al. [7]. The B. thuringiensis natural environment is thought to be the insect host intestinal system. Upon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 320 320 321 321 322 322 322 324 325 325 death of the insect the bacterium is released into the soil where B. thuringiensis is a ubiquitous inhabitant. In this environment and under favorable nutrient condition the spores could germinate and grow. The Cry genes encode the crystal toxins and are usually located on large, transmissible plasmids. The presence of Cry protein crystals in the spore is speculated to give B. thuringiensis an advantage in the soil environment upon sporulation [7] over B. cereus, B. thuringiensis is phenotypically distinguished from B. cereus only by the formation of intracellular protein crystals during sporulation. Overall, genetic studies have shown that B. cereus and B. thuringiensis are essentially identical [8]. In addition, like B. cereus, B. thuringiensis could be considered an opportunistic pathogen in animals and human [9–11]. 1.2. Bacillus cereus B. cereus is ubiquitous in nature and an opportunistic pathogen, often associated with two forms of human food poisoning, characterized by either diarrhea and abdominal distress or nausea and vomiting. In healthy individuals but mostly in individuals with certain underlying conditions such as, immunocompromised patients, or patients recovering from surgery, B. cereus has been known to cause a variety of infections, including: endophthalmitis, bacteremia, septicemia, endocarditis, salpingitis, cutaenous infections, pneumonia and meningitis [12,13]. B. cereus is found as a contaminant in many food products, including dairy products. However, its primary ecological niche is the soil environment. It is also commonly found as part of the gut microflora of invertebrates, not only as spores but also as growing vegetative cells [14]. By definition, B. cereus is acrystalliferous, but a B. cereus strain carrying a functional cry gene is considered as a B. thuringiensis strain [15]. No virulence factors specific to B. cereus have been identified, and proteins thought to be specific to B. cereus have recently been found in B. thuringiensis isolates [6]. 1.3. B. anthracis B. anthracis is the etiological agent of anthrax, an acute fatal disease found primarily among herbivores, D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 but in fact all mammals are susceptible. In recent years, it has become best known for its use as a biological weapon [16,17]. The bacterium is endemic or hyper-endemic in Africa, Asia and South America [15] but can be found in most of the world. B. anthracis spores are highly resistant to heat, ultraviolet and ionizing radiations, pressure and a variety of chemical agents and are thought to survive in the environment for decades and possibly centuries [18]. Despite the fact that B. anthracis shares the same ecological niche as B. cereus and B. thuringiensis, and can be readily isolated from contaminated soil samples, it is still unclear if its life cycle includes a vegetative stage outside the host [19]. Spores are ingested by herbivores and germinate within the host to produce vegetative cells, which multiply and produce virulence factors, ultimately killing the host. Upon death, large numbers of bacilli are released into the environment (i.e., soil), and sporulate upon contact with air, completing the life cycle. B. anthracis can be differentiated from B. cereus and B. thuringiensis though microbiological and biochemical tests. B. anthracis isolates are non-hemolytic, nonmotile, penicillin-sensitive, susceptible to c-phage, and produce a poly-c-D-glutamic acid capsule [20]. Like B. thuringiensis, the ability of B. anthracis to cause a disease is primarily attributed to its plasmid content. Fully virulent strains of B. anthracis carry two large plasmids, pXO1 (181 kb) and pXO2 (96 kb), which encode the machinery necessary to produce and regulate the anthrax virulence factors, the tripartite toxin and the capsule, respectively [21,22]. 2. Molecular identification of members of the B. cereus group of organisms: One species on the basis of genetic evidence? The classification and taxonomic separation of B. anthracis, B. cereus and B. thuringiensis has long been cause for controversy among bacteriologists, and distinguishing these species is rather difficult even with modern molecular tools. The close relationship among the different members of the B. cereus group of organisms has been established through the use of molecular techniques such as DNA–DNA hybridization [23] and more recently by comparing 16S or 23S rRNA sequences or 16S–23S rRNA spacer regions [2,24–26]. However, these methods could not adequately differentiate members of this group. There are numerous reports of attempts at developing molecular typing systems that would discriminate between B. cereus, B. thuringiensis and B. anthracis. These included multilocus enzyme electrophoresis (MEE) [3,8,27], multilocus sequence typing (MLST) [28–30], fluorescent amplified fragment length polymorphism analysis (AFLP) [31–33] and rep-PCR fingerprinting [34] among others. None of these studies 305 were able to consistently distinguish B. cereus from B. thuringiensis, but all showed a high level of genetic diversity between these two phylogenetically interspersed species. In addition, these data confirmed that B. anthracis is a monomorphic species, in which the overall diversity among isolates cannot be accurately distinguished using these techniques. Suppression subtractive hybridization was also used to catalogue some of the unique genetic content of B. anthracis [35]. Ninety-three genomic differences were discovered that could potentially be used as ‘‘signatures’’ to discriminate B. anthracis from its closest relatives but not between B. anthracis strains. The inability to separate B. anthracis isolates from one another led the Keim Genetics Laboratory to develop a robust B. anthracis molecular typing system based on variable number of tandem repeats (VNTR). A multiple-locus VNTR analysis (MLVA) using 8 then 15 loci efficiently cluster B. anthracis in two major groups (A and B), with the A group widely distributed worldwide, while the B group has a limited geographic distribution [36]. These molecular typing methods have yet to answer whether B. cereus, B. thuringiensis and B. anthracis belong to one unique species and are variants of that species. The simplest markers used for phenotypic differentiation of these species are all encoded on plasmids (i.e., the pXO2 encoded capsule gene cluster) [19]. The fact that plasmids can easily be transferred or lost makes these criteria unacceptable for typing purposes. For example, it is believed that a plasmidless isolate of B. anthracis is indistinguishable from B. cereus [37]. In addition, the isolation of B. cereus isolates containing the B. anthracis plasmid pXO1, with a novel capsule and associated with an illness resembling anthrax [20], makes this classification inappropriate. B. anthracis appears to be a highly successful variant of the B. cereus group, with a large number of strains that have been isolated, simply because it is so easy to identify cases of classical anthrax, which in turn is a readily recognizable disease [38]. Is it possible that this ease of isolation has biased our view of this group and thus B. anthracis could represent an over-sampled B. cereus? RepPCR analysis placed B. anthracis as an independent lineage in a B. cereus cluster. Perhaps all these species should be classified as B. cereus and subsequently differentiated by their plasmid content, as suggested previously [3]? Analyses of large culture collections of B. anthracis, B. cereus and B. thuringiensis isolates by AFLP and MLST [20,28,31] have identified a class of organisms containing toxigenic B. cereus and B. thuringiensis that are closely related to B. anthracis. These isolates were phylogenetically distinct from environmental B. cereus and B. thuringiensis [31] and might represent the closest ancestor to B. anthracis. 306 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 It is with this view of the genetic diversity of the B. cereus group that we have entered the genomic era. This knowledge has driven our selection of organisms to sequence and genomic-scale tools have been can be used to attempt to settle these outstanding questions. 3. Genome sequencing projects completed and in progress Like many other bacteria, the B. cereus group of organisms has benefited from the genomic revolution that started in 1995 with the publication of the first microbial genome sequence, Haemophilus influenzae [39]. Access to the genome sequence of a representative of each member of the group opens up the fields of transcriptomics and proteomics, and furthers our understanding of the mechanisms driving the specific pathogenicity of these organisms. In addition, armed with the genome sequence and a better understanding of the biology of B. anthracis, the generation of better and safer vaccine and anti-microbial drugs is possible. As described above, plasmid content confers some of the phenotypic traits used to distinguish B. anthracis, B. cereus and B. thuringiensis. Comparative genomics of members within the group can address the question – are these organisms separate species, or the same species carrying different plasmids? The first B. anthracis genome sequencing project was underway prior to the Autumn 2001 mail anthrax attacks [40,41]. This bioterrorism event has had an enormous impact on the number and choice of projects undertaken as genomic tools were applied to microbial forensics and biodefense/biopreparedness became a driver for a number of funding agencies. To date, the genome sequence of 15 isolates from the B. cereus group of organisms are available in public databases (Table 1) and others are underway. Consequently, this group of bacteria provides one of the richest collections of near neighbor sequences and will likely profoundly impact future systematics efforts. B. anthracis genome projects account for two thirds of these projects (Table 1). These 10 projects are aimed at establishing a better understanding of the genetic structure of the natural B. anthracis population, through the discovery of novel polymorphisms, and possibly of genes unique to certain strains. Single nucleotide polymorphisms (SNPs) were used to unravel the molecular evolutionary history of the group, and were the basis of a robust and finely detailed typing scheme for B. anthracis isolates [42]. The first draft genome to be published was that of the B. anthracis Ames strain isolated from the victim of the bioterror attack in Florida [42]. The choice was obviously motivated by the need to compare the draft sequence of this isolate to that of the Ames strain in progress at the time [40] at The Institute for Genomic Research (TIGR). The latter, a non-viru- lent isolate of the Ames strain was obtained from the Defense Evaluation and Research Facility at Porton Down, UK [43], where it was cured of plasmids pXO1 and pXO2 by incubation at 43 C and by novobiocin treatment, respectively [40]. The attenuated B. anthracis Ames Porton Down strain was completely sequenced and released in 2003. The original B. anthracis Ames Ancestor strain, from which both the Florida and Porton Down isolates were derived, was sequenced to completion, and is the only fully virulent strain of B. anthracis completely sequenced to date. This strain was acquired in 1981 by researchers at the United States Army Medical Research Institute for Infectious Diseases (USAMRIID) from the Texas A&M Veterinary Medicine and Diagnostic Laboratory Bacteriology Department. The strain was originally isolated from a dead cow in Sarita, TX. An original culture received in 1981 and stored at USAMRIID since, was the source of genomic DNA for sequencing. This strain is the progenitor to all the Ames strains used in laboratories around the world, hence it is an important reference. A set of six diverse isolates of B. anthracis was selected for genome sequencing to draft coverage (8·). These isolates were carefully chosen to represent as much genetic diversity as possible, as previously established through MLVA typing [36]. Three representatives of the A group (B. anthracis North Western America (AAER00000000), B. anthracis Vollum (AAEP00000000), B. anthracis Australia 94 (AAES00000000) – Table 1), two of the B group (B. anthracis Kruger B (AAEQ00000000), B. anthracis CNEVA (AAEN00000000) – Table 1) and one isolate belonging to the newly discovered C group (B. anthracis A1055 (AAEO00000000)) were sequenced and have been recently released to GenBank (Table 1). Comparative analysis of these genomes to the B. anthracis Ames Ancestor genome has provided much insight into the epidemiology and ecology of B. anthracis [42]. Lastly, the sequence of B. anthracis Sterne 34F2 (pXO1+, pXO2 ), a strain used in laboratories around the world, and cured of plasmid pXO2 by Sterne in 1937, was also released in GenBank (AE017225). It is avirulent but maintains anthrax toxin production and is the derivative of most livestock vaccines in use throughout the world [44]. B. cereus ATCC 14579 was selected for whole genome sequencing as it is non-pathogenic and is the Type strain for the B. cereus species [45,46]. The genome sequence of this B. cereus ‘‘background’’ isolate was compared to the B. anthracis Ames Florida sequence in an attempt to provide a basis for whole-genome-based phylogenetic analysis [45]. This analysis was complemented by the completion of B. cereus ATCC 10987, a nonlethal dairy isolate phylogenetically more closely related to B. anthracis than B. cereus ATCC 14579 [47]. While the genome sequence of B. cereus ATCC 14579 did reveal a small previously unknown extrachromosomal linear molecule [45], B. cereus ATCC 10987 contained a Table 1 Strains sequenced and compared in this study Alternate designation Source MLVA genotype Genotype group Plasmid genotype Coveragec GenBank Accession No. Reference B. anthracis Ames B. anthracis Ames Ancestor – A0581 Texas, Cow; plasmids cured Texas, Cow; plasmids included GT62 GT62 A3.b A3.b pXO1 , pXO2 pXO1+, pXO2+ Complete Complete [40] B. anthracis Ames Florida A2012 Patient, Florida, USA GT62 A3.b pXO1+, pXO2+ 12X GT87 B1 pXO1+, pXO2+ 12X A0071 Kruger National Park, South Africa Bison; Canada AE016879 AE017334, AE017336 (pXO1), AE017335 (pXO2) AAAC01000001, AE011190 (pXO1), AE011191 (pXO2) AAEQ00000000 GT3 A1.a pXO1+, pXO2+ 12X AAER00000000 GT79 N/Ab GT55 GT79 GT62 B2 C A3.a A4 A3.b pXO1+, pXO1+, pXO1+, pXO1+, pXO1+, 12X 12X 12X 12X Complete AAEN00000000 AAEO00000000 AAES00000000 AAEP0000000 AE017225 Brettin, et al. Unpublished, 2004 – – – – – – – – – – pXO1 , pXO2 pXO1 , pXO2 pXO1 , pXO2 pXO1+, pXO2 pXO1 , pXO2 Complete Complete Complete 12X Complete AE017194 AE016877 CP000001 AAEK00000000 AE017355 [47] [45] Brettin, et al. Unpublished, 2004 [20] Brettin, et al. Unpublished, 2004 B. anthracis Kruger B B. anthracis Western North America B. anthracis CNEVA-9066 B. anthracis A0155 B. anthracis Australia 94 B. anthracis Vollum B. anthracis Sternea A0402 – A0039 A4088 – B. B. B. B. B. – – – – – cereus ATCC 10987 cereus ATCC 14579a cereus ZK cereus G9241 thuringiensis serovar konkukian str. 97-27a Patient, Southern France Louisiana, USA Victoria Province, Australia – pXO2-deficient; the basis for animal vaccines throughout the world Dairy Dairy Zebra, Africa Louisiana, USA Severe human tissue necrosis, cf. [10] pXO2+ pXO2+ pXO2+ pXO2+ pXO2 d e f g f [41] a Not sequenced at The Institute for Genomic Research, B. anthracis Sterne and Bacillus thuringiensis serovar konkukian str. 97-27 were sequenced at The Department of Energy Joint Genome Institute, Bacillus cereus ATCC 14579 was sequenced by Integrated Genomics. b Not typed using genotyping schema to date. c Complete genomes, one contig no gaps. From GenBank – The Sterne strain of Bacillus anthracis is lacking the plasmid pXO2, resulting in an avirulent phenotype that maintains the main anthrax toxin. Prevention of anthrax in cattle can be accomplished by vaccination with living spores of the Sterne strain. Most livestock vaccines in use throughout the world are derivatives of the live spore vaccine formulated by Sterne in 1937 and still use descendants of his strain 34F2. d B. cereus ATCC 10987 harbors a large plasmid, pBc10987 (GenBank Accession No. AE017195) that is similar to pXO1, however it lacks the virulence portion that encodes the tripartite lethal toxin and associated regulatory proteins. e B. cereus ATCC 14579 contains a 15-kb linear plasmid that appears to be a phage, however it does not appear to contain any large circular plasmids. f No plasmids were released with this genomic sequence into GenBank, however plasmids present (Table 2). g B. cereus G9241 contains a plasmid that is 99.6% identical to the pXO1 plasmid from B. anthracis, however it lacks the pXO2 plasmid. There is an additional plasmid that encodes a capsule biosynthesis operon. D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 Strain 307 308 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 large circular plasmid with homology to the B. anthracis plasmid pXO1 [47]. Another group of isolates has been selected for sequencing, as they were not identifiable as B. anthracis using classical biochemical and microbiological tests as detailed above. These isolates were shown to be highly virulent and were thought to be the cause of a disease resembling anthrax in both humans and animals. The first of these atypical isolates to be sequenced was B. cereus G9241 [20], reportedly isolated from the sputum and blood of a welder with life-threatening pneumonia, whose clinical presentation resembled those of reported for 10 bioterrorism-associated inhalation anthrax patients from 2001 [17]. Interestingly, B. cereus G9241 harbors a plasmid with 99.6% identity to B. anthracis pXO1. Although homologues of pXO2-encoded capsule genes were not found, a polysaccharide capsule cluster is encoded on a second previously unidentified plasmid. B. cereus Zebra Killer (ZK) was isolated from a swab of the carcass of a dead zebra suspected of having died of anthrax in the Etosha National Park, Namibia. This isolate is phylogenetically the closest completely sequenced isolate to B. anthracis when typed by either AFLP or MLST [31] (Fig. 1). B. thuringiensis 97-27 (subsp. konkukian (serotype H34)) was originally isolated from a case of severe human tissue necrosis in a 28 year old otherwise healthy male patient [10]. Biochemical tests and the presence of crystals in sporulation culture established the isolate as B. thuringiensis. The apparent pathogenic properties of this isolate is unusual for B. thuringiensis and unlike most B. thuringiensis strains, it is closely related to B. anthracis based on phylogenetic analysis using AFLP [31]. The genome sequences of these three isolates open the possibility of developing a better understanding of their pathogenic potential when compared to B. anthracis. These sequences may also provide a snapshot of the closest phylogenetic ancestors to B. anthracis. However, these strains are the exception rather than the rule, and may instead constitute another successful evolutionary lineage of B. cereus. Despite its importance as a biopesticide, B. thuringiensis as a non-pathogenic species is under-represented in the collection of whole genome sequences. A single genome, B. thuringiensis subsp. israelensis, has been sequenced to 8· and assembled in more than 800 contigs (www.integratedgenomics.com), but is not available to the scientific community for analysis. However, the sequence of the crystal toxin-encoding plasmid pBtoxis has been recently published [48] and showed some similarity with B. anthracis plasmid pXO1. The isolates for which whole genome sequences are now available do not properly represent the diversity observed in the B. cereus group of organisms using methods such as MLST (Fig. 1). The choice of strains sequenced so far reflects a bias driven by a need to understand rare pathogenic traits in some of these species. Two non-pathogenic B. cereus are available to the public and one non-pathogenic B. thuringiensis isolate has been sequenced, while 13 pathogenic isolates have been sequenced. The high number of B. anthracis strains Fig. 1. Multi-locus sequence typing (MLST) neighbor-joining phylogenetic tree of representatives of the B. cereus group of organisms. All B. anthracis isolates (ST1-3) colored red are represented as a single point on the tree. B. cereus isolates are colored black and B. thuringiensis are colored in blue. ‘‘ST’’ following each strain designation indicates the sequence type as listed in the PubMLST database [29]. Isolates that have been sequenced are boxed in gray. D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 sequenced has taken the whole genome sequencing approach to the limits of its resolution and has led to a phylogenetic resolution of the group never achieved for any other bacterial species [42]. 4. B. anthracis genomic analysis for molecular and forensic epidemiological purposes The disciplines of emerging infectious disease and forensic microbiology have shifted from using biological phenotypes as markers to using more reliable and quantifiable molecular markers, such as SNPs [49]. B. anthracis, as shown above, has received considerable attention because of its demonstrated use as a biological weapon and the difficulties associated with forensic tracking of this genetically homogenous group of organisms [36,42]. In response to the threat inherent to B. anthracis, vast resources have been allocated to develop diagnostic characters for this species through genome sequencing. Soon after the Autumn 2001 attacks [50], a draft genome sequence of a B. anthracis Ames Florida strain was generated, and compared to the nearly complete genome sequence of the plasmid-less B. anthracis Ames Porton strain [41]. This analysis revealed 60 new markers that included SNPs, indels and tandem repeats. Only four differences were discovered between the main chromosomes of the Florida and Porton isolates (two SNPs and two short indels). For the first time polymorphic markers discovered through comparative genome sequence analysis were used to test a collection of anthrax isolates and were able to divide these samples into distinct subgroups [41]. This study was critical in establishing that infectious disease outbreaks, both naturally occurring and maliciously released (bioterrorism), could be investigated through whole genome-based analysis. More importantly, the study introduced statistical models allowing scientists to discriminate true polymorphism from random sequencing error. This was critical as in light of the rapid generation of B. anthracis draft genome sequences. A draft genome is by definition unfinished, and the poor quality of portions of a draft sequence is of little use for polymorphism discovery. The set of polymorphisms found when these two strains were compared was not sufficient for typing unsequenced strains. It was speculated that sequencing other distantly related B. anthracis strains might yield additional polymorphisms and increase the resolution of the method. A recent study applied this approach to a larger number of B. anthracis draft genomes and discussed the phylogenetic discovery bias that resulted from using SNPs extracted from whole-genome sequence comparisons [42]. More than 3500 rare high quality SNPs were discovered when the genome of five B. anthracis draft genomes were compared to the closed B. anthracis Ames. 309 A sub-set of 990 SNPs were then typed against a panel of 26 diverse B. anthracis strains using a high-throughput assay based on the SnaPshot primer extension protocol (Applied Biosystems). This data demonstrated that precise phylogenetic topologies could be achieved, yielding accurate information on internodal distances. In addition, using appropriate B. cereus outgroups, the authors were able to determine the evolutionary root of the B. anthracis clades. The root lies closer to a newly described group C than either of the two previously described A and B groups. Unequal evolutionary rates were observed among the sequenced isolates that could be correlated with ecological parameters and strain attributes, such as host availability in its natural environment. The young evolutionary characteristics of the B. anthracis group were confirmed by the rarity of SNPs and low overall homoplasy. Armed with this new set of markers, it is now possible to select ‘‘canonical SNPs’’ for identifying long branches or key phylogenetic positions [51]. A molecular typing strategy that maximizes the use of markers discovered through whole-genome comparison was recently established for B. anthracis [51]. PHRANA (Progressive hierarchical resolving assays using nucleic acids) is a nested approach that employs canonical SNPs, MLVA (15 loci), and simple nucleotides repeats (SNRs). PHRANA makes use of both the low resolving power of canonical SNPs and the high resolving power of SNRs, in a sequential manner. This approach allows for high-resolution and accurate representation of the relationships between B. anthracis isolates by minimizing the effect of homoplasy using a stepwise approach [51]. 5. Comparison of the chromosome sequences of the B. cereus group of organisms 5.1. Non-pathogenic B. cereus ATCC 14579 and B. cereus ATCC 10987 The genome sequences of two non-pathogenic B. cereus isolates (ATCC 14579 and ATCC 10987) have been completed and compared to that of B. anthracis [45,47]. To better understand the degree of relatedness of members of the B. cereus group, one should compare the chromosome gene content and genetic structure. These organisms demonstrate a wide range of phenotypes and pathological effects, however these effects are often derived from factors encoded on extra-chromosomal elements, such as the large toxin encoding plasmid pXO1 in B. anthracis. Sequence analysis of nine chromosomal genes has suggested that B. anthracis, B. cereus and B. thuringiensis should be considered as one species [3]. Access to the complete genome sequence of both B. cereus and B. anthracis isolates gave the opportunity to revisit the question and explore the genetic content that governs some of these specific phenotypes. 310 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 A detailed comparative analysis of the genomes of B. cereus and B. anthracis, revealed a small subset of genes unique to either species, most of which are annotated as hypothetical [47]. The majority of these genes are located at the terminus of replication, indicating that genome plasticity mostly occurs in that region, as previously observed for other microbial groups [52]. These data suggest a history of insertion and/or deletion in the evolution of the B. cereus group [47]. It was observed that, in many cases, genes found at a specific position in one genome were replaced with others at the corresponding loci in another. These regions often were the result of insertion/deletion events of mobile genetic element such as phages, transposons, IS elements or metabolic adaptations. For example, a nine-gene urease gene cluster was identified in the genome of B. cereus ATCC 10987 that replaces the B. anthracis/B. cereus ATCC 14579 genes that encodes for hypothetical proteins, blasticidin S deaminase and S layer protein. Additionally, a xylose utilization operon was found to replace functions such as nitrate reduction, nitrite reduction and molybdopterin synthesis. The lack of nitrate and nitrite reduction appears to have been compensated by the acquisition of the urease operon, allowing the bacterium to use ammonia as a nitrogen source derived from urea. In addition, B. cereus ATCC 10987 contains a unique set of genes responsible for the transport and utilization of tagatose. This particular strain has been isolated during a study on cheese spoilage, where this carbohydrate can be found. This replacement might represent another example of metabolic adaptations to the carbohydrate-rich environment of milk [47,135]. A cluster encoding for the arginine deiminase pathway was identified in both B. cereus ATCC 10987 and ATCC 14579 that, like in Streptococcus pyogenes, might enable B. cereus to survive in acidic conditions [45]. On the other hand, it is thought that ammonium inhibits receptor-mediated internalization of the lethal toxin, hence deletion of the arginine deiminase pathway in B. anthracis might be the result of an evolutionary adaptation to a pathogenic lifestyle [45]. These specific evolutionary events represent niche specific adaptations, but do not eliminate the fact that they are highly similar and syntenic (conserved gene order). An analysis using normalized blast scores to compare the proteomes of B. cereus G9241, B. cereus ATCC 10987 to that of B. anthracis, demonstrated how syntenic these three genomes are [47,135]. Fig. 2 represents a similar analysis with an added level of color-coded protein similarity and shows the high level of gene similarity and gene order (Figs. 2(a) and (b)), with no inversions or genetic rearrangements of large genome segments. This analysis also shows that B. cereus G9241 is more closely related to B. anthracis than to B. cereus ATCC 10987 (Fig. 2(c)). These bioinformatic results are mirrored in the increased pathogenicity of B. cereus G9241. Using a B. anthracis DNA microarray, comparative genome hybridization (CGH) was performed with 19 diverse isolates from B. cereus group of organisms, including B. weihenstephanensis [40]. CGH confirmed the overall similarity of chromosomal genes among this group of close relatives. Interestingly, the set of core genes conserved across each members of the group is located around the origin of replication, This study also revealed six regions unique to the B. anthracis chromosome, comprising four phages regions and IS110 related insertion elements. Not surprisingly, the presence of non-toxin related pXO1 gene homologues were detected in half the 19 strains tested and is further evidence for the mobility of pXO1 genes within the B. cereus group (see section below). In contrast, there were few pXO2 gene homologues found in the set of isolates tested. Chromosomally encoded genes that may contribute to pathogenicity, including hemolysins, phospholipases and iron acquisition systems were identified in the genomes of B. anthracis. These include genes similar to those known to boost viral infectivity by degrading the mucin layer of insect guts, and genes that contribute to the virulence of the Gram-positive pathogen, Listeria monocytogenes [40]. These genes and some newly identified surface proteins constitute potential targets for drug and vaccine development. Analysis of the metabolic potential of both B. anthracis and B. cereus shows that these organisms have an expanded capacity for amino-acid and peptide utilization, and hence are equipped for a life in a protein rich environment. This lifestyle was emphasized by the presence of six amino-acid efflux systems, which prevent accumulation of amino acids to bacteriostatic concentrations during growth on peptides [40]. In addition, complex carbohydrate degradation appears to be favored by these organisms over small sugars. They indeed lack catabolic capacity for the utilization of mannose, arabinose and rhamnose among others, but seem to be capable of cleavage of extracellular chitin and chitosan [40]. These metabolic capabilities suggest that the most recent ancestor to the B. cereus group of organisms might have been able to thrive on animals or insects [53]. Comparative analyses suggest that major differences between members of the B. cereus group might represent fine alteration in gene expression rather than the level of sequence divergence or gene content [40,45,47]. PlcR is a pleiotropic transcriptional regulator that upregulates the expression of more than 100 genes in B. cereus through binding to an upstream palindromic motif [45,54,55]. PlcR activity has been shown to be regulated by the presence of a secreted and reimported pentapetide produced from the processing of the PapR protein C-terminus [56]. The papR gene is itself upregulated by PlcR, forming a quorum sensing-like system [56,57]. Proteomic-based comparative analysis of B. cereus ATCC 14579 and a DPlcR D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 311 Fig. 2. Blast score ratio analysis of B. cereus G9241, B. cereus ATCC 10987 and B. anthracis Ames Ancestor. Blast score ratios are obtained by dividing the Blast score for the most similar query peptide versus the reference peptide by the Blast score for the reference peptide against itself. The Blast score ratio can then be graphically represented as a synteny plot of two genomes using the genomic location of the best hit in each genome as the coordinates on the Cartesian plane [135]. These synteny plots demonstrate that pathogenic and non-pathogenic B. cereus group members have a high degree of similarity as indicated by the color of each peptide, represented by a dot, and conserved gene order (synteny) represented by the dots forming a line with a slope of 1. (a) Synteny plot of B. anthracis vs. B. cereus G9241. (b) Synteny plot of B. anthracis vs. B. cereus ATCC 10987. The scale at the top of the figure indicates the color and Blast score ratio correlation represented in (a) and (b). A Blast score ratio of less than 0.4 is not considered significant. (c) Blast score ratio analysis of B. cereus G9241 and B. cereus ATCC 10987 using B. anthracis Ames Ancestor as the reference. The Blast score ratio for each of the query genomes can also be used to obtain an overall idea of similarity between the genomes compared (red – highly conserved in all three genomes; orange – unique to the genome used as the reference; green – shared between B. anthracis Ames Ancestor and B. cereus G9241; blue – shared between B. anthracis Ames Ancestor and B. cereus ATCC 10987). 312 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 variant of the same strain by two-dimensional gel electrophoresis has identified collagenase, phospholipases, hemolysins, proteases and enterotoxins genes that are PlcR upregulated [54]. Some of these activities are considered virulence factors in B. cereus and all of their associated genes displayed a PlcR binding box in the promoter region. Even though these genes are also found in B. anthracis, a nonsense point mutation in the PlcR gene is responsible for an abolition or dramatic reduction in their expression such that production of lecithinase, protease and hemolysins is undetectable [40]. A few genes encoding cytotoxin K and non-hemolytic enterotoxin C subunit, amongst others, have only been found in B. cereus [47]. The acquisition of the toxin encoding pXO1 plasmid and its regulator AtxA has been shown to be incompatible with the chromosomally encoded PlcR [58]. Interestingly, a recent study showed that a fused PlcR–PapR construct was able to restore strong hemolytic activities when introduced in B. anthracis [59], indicating that these genes, while not upregulated, are still fully functional and have not undergone evolutionary decay. The loss of regulation of these chromosomally encoded genes might represent another example of an adaptive response of B. anthracis to a plasmid driven advantageous pathogenic lifestyle [40] and because these genes are still fully functional, it confirms the relatively young evolutionary age of B. anthracis. 5.2. The pathogenic members of the B. cereus group of organism, B. cereus G9241, B. cereus Zebra Killer, and B. thuringiensis 97-27 The genome sequences of one B. thuringiensis and two B. cereus isolates that were associated with severe disease have been sequenced in order to gain insight into their pathogenic potential. While one can only infer the pathogenicity of B. cereus ZK based on its isolation from a dead zebra, the opportunistic pathogenicity of B. thuringiensis 97-27 has been confirmed by infection of both immunosuppressed and immunocompetent mice [9–11]. On the other hand, the pathogenic characteristics of B. cereus G9241 are supported by the severe anthraxlike clinical presentation it caused. This isolate has been shown to be lethal in A/J mice challenged by intraperitoneal injections [20]. The genome sequences of these isolates give us the opportunity for comparison to the non-pathogenic B. cereus isolate and B. anthracis. Structurally, these genomes show a high level of synteny and protein identity to B. anthracis and non-pathogenic B. cereus isolates. Overall, the proteome of B. thuringiensis 97-27, B. cereus ZK and B. cereus G9241 show a higher similarity to that of B. anthracis than to that of the non-pathogenic B. cereus ATCC 14579. These observations are in agreement with the phylogenetic position of these isolates relative to B. anthracis inferred using AFLP [31] or MLST ([28] and Fig. 1). In common with other members of the B. cereus group, B. cereus G9241, B. cereus ZK, and B. thuringiensis 97-27, share a set of genes that are associated with virulence, such as non-hemolytic enterotoxins, channel-forming type III hemolysins, a perfringolysin O (listeriolysin O), phospholipases C, and a family of extracellular proteases [60]. Interestingly, the hemolytic B. cereus ZK does not contain the hemolysin BL (hbl) operon, which is a primary factor in diarrheal B. cereus food poisoning [61]. In contrast, B. cereus G9241 contains four hemolysins (A, BL, II and III), and has also been shown to be phenotypically hemolytic [16]. In line with other B. cereus and B. thuringiensis isolates, these strains contain a plcR gene encoding for a full-length, potentially functional protein and a similar set of genes putatively upregulated by this protein, with a few exceptions. The histidine protein kinase (BC3528) homologous to the sporulation kinase KinB is a member of the PlcR regulatory network in B. cereus ATCC 14579 [45] and B. cereus G9241 [20], but is absent in B. cereus ATCC 10987 [47], B. anthracis, B. thuringiensis 97-27 and B. cereus ZK. The genomes of these pathogenic members of the B. cereus group also contain metabolic adaptations similar to those found in the non-pathogenic group. For example, like B. cereus ATCC 10987, B. cereus ZK contains a 14 kb gene replacement consisting of a gene cluster responsible for the transport and utilization of tagatose [47]. Similarly, B. thuringiensis 97-27, but not B. cereus G9241 or B. cereus ZK, is able to utilize arginine through the arginine deiminase-dependent pathway, which is also found in B. cereus ATCC 14579 and B. cereus ATCC 10987 [45,47]. These metabolic adaptations certainly reflect the environment in which each of the organisms thrive, and does not appear to be link to a pathogenic characteristic of any of these isolates. Interestingly, and relevant to its pathogenic potential, a putative polysaccharide capsule cluster similar to the one present on B. cereus ATCC 14579 is also present on B. thuringiensis 97-27. While a distantly related polysaccharide capsule cluster is found in the same genomic location in B. cereus ATCC 10987 no polysaccharide capsule cluster is found on the genomes of either B. anthracis or B. cereus ZK. Unfortunately, no experimental data are available that demonstrate B. cereus ZK or B. thuringiensis 97-27 encapsulation in vivo or in vitro. In contrast, B. cereus G9241 has been shown to produce a capsule by staining with India ink [20] and a novel polysaccharide capsule gene cluster has been identified on one of its plasmids. However, no other identifiable capsule biosynthetic gene cluster has been identified in the genome sequence. Comparative analysis of the genome of these pathogenic isolates do not conclusively provide the informa- D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 tion necessary to assign a particular set of genes to their virulent phenotype. Many of the differences are shared with other non-pathogenic counterparts. It appears that B. cereus ZK shares the most similarity with B. anthracis, justifying AFLP typing data as the closest phylogenetically isolate to B. anthracis [31]. These isolates all contain small and/or large plasmids, for which sequence is now available. A detailed analysis of these plasmids might provide a better insight into the pathogenicity of their host isolates. 6. The B. cereus group of organisms plasmid sequence comparisons Historically, pathogenic potential and diverse host range have defined the members of the B. cereus group. The small, usually metabolic-based differences found in the chromosomal content cannot account for the spectrum of disease and host range observed in this group. However, much of the disease and host specificity in this group can be attributed to plasmid content. The plasmids associated with the B. cereus group of organisms have broad size range (5–200 kb) and vary in number ([62] and Table 2). Questions about their role in the life cycles of the species that harbor them remain. The most striking example of the plasmid content affecting host range and pathogenesis occurs in B. anthracis. B. anthracis harbors two plasmids, one that encodes the tripartite lethal toxin complex, pXO1 [22] and the other, which contains the biosynthetic genes for the poly-c-D-glutamic acid capsule [21,63]. While each plasmid is a distinct entity, it has been shown that loss of either one results in an attenuated B. anthracis isolate. The role of the B. anthracis plasmids in pathogenesis is exquisitely known, how- 313 ever, the function of the other plasmids in the group is relatively unknown. However, one could expect that these plasmids encode the peptides responsible for the specific phenotypes that differentiate the members of this group. 6.1. General features of the B. cereus group plasmids identified in genome sequencing 6.1.1. B. anthracis plasmids 6.1.1.1. pXO1. The 181-kb plasmid pXO1 encodes genes for the production and regulation of the tripartite lethal toxin. The three portions of the lethal toxin are produced separately and assembled. The eukaryotic membrane-interacting protective antigen (PA) is expressed as a pre-protein and proteolytically processed into its active form, known to elicit a protective immune response against anthrax [64]. The toxin has two additional components – the lethal factor (LF) and the edema factor (EF), which are responsible for the proteolytic cleavage of several mitogen-activated protein kinases (MAPKKs) and convert intracellular ATP into cAMP, respectively. The biological activities of these subunits are not fully characterized but result in aberrant signaling inside the macrophages and fluid accumulation in the lung [65]. All three subunits are required for active toxin production and activity (for reviews see [66–70]). The three structural genes, cya (EF), lef (LF) and pagA (PA), are under the control of at least two regulatory elements, AtxA and PagR [71], and have been shown to be expressed early in the growth of B. anthracis [72]. These genes are encoded within a 44.5-kb pathogenicity island that is transpositionally active [19]. However, its inversion does not appear to affect virulence [41]. Table 2 Comparison of plasmid sequences from plasmids identified in genome sequencing projects Plasmid name Organism Strain Size (bp) G+C (%) Replicon Replication system Number of CDS Functional CDS Hypothetical CDSa pXO1 pXO2 pBc10987 pBClin15 pBCXO1c pBC218d pZK5 pZK8 pZK9 pZK54 pZK467 pBT9727 B. B. B. B. B. B. B. B. B. B. B. B. Allb Allb ATCC 10987 ATCC 14579 G9241 G9241 Zebra Killer Zebra Killer Zebra Killer Zebra Killer Zebra Killer 9727 181,677 94,830 208,369 15,100 191,110F 218,094F 5108 8191 9150 53,501 466,370 77,122 33 33 33 38 33 32 31 32 31 32 33 33 Unknown pAMb1 Unknown Phage Unknown Unknown Group VII Group VII Group VII pAMb1 Unknown pAMb1 Unknown Theta Unknown Phage Unknown Unknown RCR RCR RCR Theta Unknown Theta 204 104 242 21 177 185 5 8 10 58 465 80 48 26 91 4 61 116 2 4 5 27 228 18 156 78 151 17 116 69 3 4 5 31 237 62 a anthracis anthracis cereus cereus cereus cereus cereus cereus cereus cereus cereus thuringiensis Hypothetical includes the proteins with no know function including CDS with hypothetical, conserved hypothetical and Bacillus cereus groupspecific protein designations. b B. anthracis pXO1 from GenBank Accession No. AE017336. B. anthracis pXO2 from GenBank Accession No. AE017335. c GenBank Accession No. AAEK01000020, AAEK01000036 and AAEK01000038 (not currently closed molecules). d GenBank Accession No. AAEK01000004. 314 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 Within the isolates of B. anthracis for which the genome sequence is available, the pXO1 sequences are highly similar. Apart from a small number of SNPs, SNRs and VNTRs, no large insertions or deletions have been observed [41]. To date only one B. anthracis isolate has been identified without the pXO1 plasmid [42]. This isolate represents a new lineage in the B. anthracis group. However, little information is available for this isolate and it is possible that pXO1 might have been lost during laboratory passages. The level of sequence coverage obtained for pXO1 from whole-genome shotgun sequencing projects can be used to estimate the molecular ratio of plasmid to chromosome [41]. For pXO1, the copy number has been suggested to be 2–3 copies per chromosome copy ([41] and this study). This estimate appears to be much higher than indicated by other studies [73], and certainly represents a snapshot of the dynamic plasmid content in the bacterium at the time of genomic DNA preparation. The origin of replication of pXO1 has never been conclusively identified [22,74]. It has been suggested that it lies within an 11-kb region discovered by subcloning [22,75], however this region does not contain any genes similar to other known plasmid replication systems. This is a recurring theme among the large plasmids of the B. cereus group and is addressed further below. While many studies have focused on the toxin and its regulators, five years after the complete sequence of pXO1 was first published [22], 148 of the 204 potential coding sequences (72.5%) remain functionally unidentified. Detailed transcriptional mapping studies should be able to refine the annotation and in the process identify novel genes involved in the pathogenicity of the organism in the future. 6.1.1.2. pXO2. pXO2 (96 kb) encodes for the synthesis and degradation of another well-known virulence factor, the poly-c-D-glutamic acid capsule [76]. This capsular material is thought to protect the vegetative bacterial cells during transit through the macrophage, hence evading the host immune response and increasing systemic sepsis [77]. The poly-c-D-glutamic acid capsule is produced by a three-gene operon capABC under the control of a number of regulators including the toxin regulator AtxA encoded on pXO1 [71], and the pXO2 encoded regulators AcpA and AcpB. Degradation of the capsular material, by CapD [78], encoded on pXO2, results in high and low molecular weight forms of the capsule, both of which essential for infection [79]. B. anthracis Sterne lacks pXO2, and is commonly used around the world as an anthrax veterinary vaccine. Very little is known about the molecular mechanism for the transport of the capsular material to the surface of the bacterial cell. The first complete sequence of pXO2 was obtained from a B. anthracis Pasteur strain (pXO1 , pXO2+) [21] and encodes 104 genes for which 78 do not have functional assignments (78/104, 75%). In contrast to pXO1, the pXO2 replication region has been identified and, recently, a 60-nucleotide region essential for the initiation of plasmid replication has been characterized [80]. pXO2 is a theta replicating plasmid similar to the prototypical pAMb1 plasmid from Enterococcus feacalis. Detailed analysis of the replication machinery is provided below. Several studies have indicated that pXO2 sequences are not widely distributed among closely related species based on CGH, hybridization and PCR experiments [40,63]. Only one isolate, B. thuringiensis AW06, showed any significant sequence similarity to pXO2 in previous studies, however the B. thuringiensis plasmid pBT9727 described in this work has significant similarity to pXO2 (see below). 6.1.2. B. cereus plasmids In contrast to the conserved plasmid content observed among B. anthracis, B. cereus isolates contain a diverse range of plasmids – no strains have yet been identified with identical plasmid content. B. cereus plasmids vary from 5- to almost 500 kb in size and only a limited number have been implicated in pathogenesis (Table 2). A subset do not encode for any obvious phenotypes and may be considered cryptic. B. cereus genome projects have identified a large number of plasmids, and their analysis has revealed a number of conserved regions within the large plasmids group. The smaller plasmids, in contrast, show greater similarity to B. thuringiensis smaller plasmids. 6.1.2.1. pBc10987. A single 208-kb plasmid, named pBc10987, was identified from the non-pathogenic isolate B. cereus ATCC 10987 [47]. pBc10987 shows surprising similarity to the plasmid pXO1 of B. anthracis (40% nucleotide identity), however it lacks the pathogenicity island (PI) containing the genes that encode for the tripartite lethal toxin and its associated regulators [47]. In lieu of the pXO1 PI, pBc10987 contains genes potentially involved in adaptation to either an environmental or pathogenic lifestyle. Environmental adaptations of pBc10987 include a copper requiring tyrosinase, arsenite resistance and its associated regulators as well as an amino acid transport system. The pathogenic adaptations comprise of two potential novel toxins [22,47]. Unlike the PI in pXO1, this region in pBc10987 is not flanked by mobile genetic elements, suggesting that it has not been lost from pBc10987 [22,47]. These findings suggest that in conjunction with the chromosome, B. cereus group plasmids contribute to the metabolic fitness of the species. In addition to these potential metabolic or pathogenic adaptations, a number of aspects of plasmid biology and chromosome-plasmid crosstalk are conserved between pXO1 and pBc10987. Like pXO1, the replica- D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 tion machinery of pBc10987 has not been identified using bioinformatics tools, however the similar plasmid copy number (1–3 copies per cell) suggests that the replication mechanism is conserved between the two. Divergent copies of abrB, a pleiotropic transition state regulator, are present on pXO1 and pBc10987, as well as the chromosomes of B. anthracis and B. cereus ATCC 10987. AbrB has been shown in B. subtilis to modulate the switch between biofilm formation and sporulation [81], as well as regulating competence [82,83]. In B. anthracis, AbrB has been shown to negatively regulate toxin production [84,85]. While pBc10987 and pXO1 AbrB proteins are similar, they differ significantly at their N-terminus, in that the pXO1 protein is 27 amino acids shorter rendering it potentially inactive [84]. The role of the pBc10987 encoded AbrB protein is currently unknown, but it is speculated that it acts as a regulator of the genes replacing the pXO1 pathogenicity island [47]. Besides AbrB, there are two more examples of possible genetic exchange between the chromosome and the plasmid of the B. cereus ATCC 10987. Identical copies of a transposable element similar to the S. aureus Tn554 [86] are present on both the chromosome and plasmid. There are four genes associated with the Tn554-like element and one, bclA, is of interest. In B. anthracis, multiple functions have been attributed to BclA. It is the major spore surface antigenic protein [87,88] and has been shown to play a role in the depth determination of the spore coat [89]. Divergent proteins of the same family as BlcA have been found on the chromosomes of B. anthracis and some plasmids of B. thuringiensis [90]. Interestingly, the bclA gene in B. anthracis is limited to the chromosome, whereas in B. cereus ATCC 10987 similar copies of bclA are present on the chromosome and the plasmid. The presence of similar genes on other plasmids suggests that genetic exchange occurred between plasmids and chromosomes in the B. cereus group of organisms. 6.1.2.2. pBCXO1 and pBC218. B. cereus G9241 has been associated with an illness resembling inhalation anthrax [20]. Initial CGH studies revealed that B. cereus G9241 contained genes with a high degree of hybridization to pXO1, including all three anthrax toxin genes, however, no hybridization was observed to pXO2. Unexpectedly, a 191-kb plasmid with a high degree of similarity and synteny to B. anthracis pXO1 was found in B. cereus G9241 [20]. In addition, B. cereus G9241 contains a second 218-kb plasmid, previously unidentified, that encodes a novel polysaccharide capsule biosynthetic cluster. These plasmids are expected to contribute greatly to the observed anthrax-like clinical presentation. The pXO1-like plasmid, named pBCXO1, is 99.6% identical to pXO1. This similarity extends to the pXO1 315 virulence genes, protective antigen (99.7% amino acid identity), lethal factor (99% amino acid identity) and edema factor (96% amino acid identity), as well as the known virulence regulatory proteins AtxA (100% amino acid identity) and PagR (98.6% amino acid identity) [20]. Price et al. [91] have previously used minor variations in the protective antigen (PA) protein as a classification system. Using this system, pBCXO1-encoded PA is most similar to genotype V, often associated with the western North America diversity group of B. anthracis [20]. ELISA experiments demonstrated that PA was present in the supernatant of a stationary phase culture, however it is unclear which protective antigen subunit was recognized by this assay [20]. This high level of identity between pXO1 and pBCXO1 suggests that both isolates acquired their plasmid from a common ancestor or that transfer occurred into B. cereus G9241 from B. anthracis. The successful transfer of pXO1 from B. anthracis to a close relative with the aid of a mobilizing plasmid [92] supports the latter. The regulators, PlcR and AtxA, have been shown to be functionally incompatible in B. anthracis. The introduction of pXO1-encoded AtxA is thought to have led to the selection for the plcR nonsense mutation in B. anthracis [58]. Interestingly, B. cereus G9241 appears to encode fully functional copies of both PlcR and AtxA. However, PlcR contains a region of lower similarity to B. anthracis PlcR that is thought to compensate for the presence of AtxA or represent another potentially inactive form of PlcR. It is also hypothesized that the pBCXO1 plasmid may have been acquired recently and the incompatibility has not had time to generate a nonsense mutation. In light of the discovery of this important organism, functional studies of B. cereus G9241 regulatory pathways must be performed to develop a better understanding of this evolutionary event. B. cereus G9241 contains a second plasmid, pBC218 (218 kb), with limited similarity to a region in pZK467 (Fig. 3(b)). Intriguingly, pBC218 carries a second copy of atxA, however the gene product is only 78% identical to the B. anthracis homologue [20]. In addition, pBC218 encodes some of the known B. anthracis virulence factors [20]. Genes encoding for a homologue of the protective antigen peptide (60% amino acid identity) and the lethal factor (36% amino acid identity) are also found on pBC218, however the plasmid does not contain a homologue of the edema factor. The lethal factor is significantly truncated when compared to that of B. anthracis pXO1 and is probably not functional. In contrast, the pBC218-encoded protective antigen peptide is identical to B. anthracis at all 10 dominant negative amino acid residues identified by Mourez et al. [93]. Another 33 residues were identified that resulted in decreased activity when substituted by cysteine, of which 27 are identical in pBC218-encoded protective antigen, with the remaining six being 316 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 viously thought that B. cereus, as a species in the group, did not produce capsule [6]. Further examination of the pBC218 sequence revealed a putative polysaccharide biosynthetic operon, representing the only capsule biosynthetic cluster identified in the genome of B. cereus G9241. This gene cluster is thought to be responsible for the capsule produced by this isolate. The lack of any other capsule suggests that this plasmid-encoded polysaccharide capsule might compensate for the lack of poly-c-D-glutamic acid capsule and help B. cereus G9241 to evade the host immune system. The plasmid content of this isolate represents a composite of B. cereus and B. anthracis. It also raises interesting questions regarding the regulatory interaction between AtxA and PlcR, the assembly of the toxin and the role of the novel capsule in pathogenesis. Fig. 3. Identification of regions with proteomic and syntenic similarities by comparisons of selected B. cereus group plasmids. Blast score ratio analyses were used to compare the putative plasmid proteomes as described in Fig. 2. (a) Synteny plot of pBT9727 vs pXO2. The pBT9727 plasmid contains a similar backbone structure to pXO2. The replication proteins are conserved in plasmid location and peptide sequence however pBT9727 lacks the poly-c-D-glutamic acid capsule biosynthetic genes, indicated by the gray box. An additional unique region of 10 kb is identified in pBT9727 by the gray box (22–33 kb). 48 of the 80 – pBT9727 peptides have homologues in pXO2. (b) Comparison of pBC218 and pZK467 demonstrates that these plasmids have limited similarity with the exception of one region, highlighted in the gray box. This region is conserved in both gene content and gene order. conservative substitutions [20,93]. The presence of these additional toxin subunits is raising some interesting questions about their putative function in the pathogenesis of this organism. Could the pBC218-encoded PA form a complex with the pBCXO1 lethal factor and edema factor to form what would appear to be novel functional anthrax toxin? How do these fragments interact and are they coordinately regulated? India ink staining and microbiological analyses demonstrated that B. cereus G9241 is encapsulated and that capsule production is not regulated by increased CO2 concentrations like the B. anthracis pXO2-encoded poly-c-D-glutamic acid capsule. Interestingly, It was pre- 6.1.2.3. pBClin15. A 15-kb linear plasmid, pBClin15, was identified by genome sequencing of B. cereus ATCC 14579 [45]. This plasmid contains none of the typical replication machinery associated with small plasmids identified in either B. cereus or B. thuringiensis, but does contain genes that suggest that it may represent a novel phage. Recently, it has been reported that pBClin15 shows some level of similarity to the Bam35 phage of B. thuringiensis var. alesti strain 35 [94,95]. Additional comparisons and experimental data suggest that Bam35 can exist both as a prophage and a free phage [95]. In contrast to Bam35, pBClin15 lacks terminal inverted repeats that may allow protein-primed replication, an observation that led to the suggestion that pBClin15 is a degenerate phage [95]. 6.1.3. B. cereus Zebra Killer (ZK) plasmids The plasmids from B. cereus ZK represent a novel set of plasmids carried by an isolate that causes a disease resembling anthrax in animals (P.C.B. Turnbull, personal communication). Five distinct plasmids were identified in the genome of B. cereus ZK, representing the largest number of plasmids harbored by a strain of B. cereus so far. Interestingly, the three small plasmids (less than 10 kb, Table 2) pZK5 (CP000041), pZK8 (CP000043) and pZK9 (CP000044) are more similar to known B. thuringiensis plasmids than any previously sequenced B. cereus plasmids, whereas the larger two plasmids, pZK54 (CP000042) and pZK467 (CP000040), share characteristics with pXO2 and pBC218, respectively. The three small plasmids are predicted to replicate through a rolling-circle mechanism; their functional annotation is mostly limited to the identification of replication and mobilization proteins. pZK54 (54 kb) encodes a pXO2-like putative theta replication system, however no identifiable replication system was found in pZK467 (466 kb), as in pXO1 [17,76]. pZK467 and pBC218 share an approximately 40-kb region where synteny is retained, however, no functional D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 assignments exist in this region (Fig. 3(b)). pZK467 is the only known plasmid with similarity to pBC218. A large number of transposase genes or mobile elements have been identified in these two large plasmids. These elements may facilitate gene exchange between plasmids as well as between plasmids and the chromosome. None of the B. cereus ZK plasmids encode homologues of known virulence factors in B. anthracis, B. cereus or B. thuringiensis [6]. Sequence analysis of B. cereus ZK plasmids does not improve our understanding of the pathogenicity of this organism, which currently remains unclear. Analysis of the relative copy number of these plasmids obtained from sequence analysis indicates that each has a copy number of 1–2, except for pZK5 which is estimated at 0.6. These data might suggest that pZK5 is unstable under the laboratory culture conditions employed and is in the process of being lost. 6.1.4. B. thuringiensis plasmids Phenotypic characterization of B. thuringiensis is often based on the presence of plasmid-encoded large Cry protein inclusions of the d-endotoxin [48]. Recently, a study by Andrup et al. [90] described six B. thuringiensis plasmids (less than 20 kb) that did not carry the cry gene. It is possible to group B. thuringiensis plasmids based on the similarity of their replication and mobilization machinery, however this system did not extend to the larger B. thuringiensis or B. cereus plasmids. 6.1.4.1. pBtoxis. In addition to the four known Cry and two known Cyt toxins, the 128-kb circular plasmid, pBtoxis encodes a third Cyt-type sequence with an additional C-terminal domain previously unseen in such proteins [48]. B. thuringiensis subsp. israelensis carries pBtoxis, and its toxin crystals have been demonstrated to be one of the most toxic combinations tested [96]. However, it is unclear if this toxicity is related to the specific combination of toxins or to toxin interaction with other plasmid encoded features. GC skew analysis indicated a putative origin of replication, however no replication proteins has been identified, similar to the other large B. cereus group plasmids (Table 2). The coding sequences adjacent to this region in pBt001 showed >78% amino acid identity to pXO1-49, and is located near a similar putative replication origin on pXO1, also predicted by GC skew analysis. However, experimental functional evidence is not available. In addition to the toxin genes, pBtoxis encodes a number of genes that are thought to enhance crystal formation and subsequent cell viability by acting as chaperones. Interestingly, like pBC10987 and pXO1, pBtoxis encodes peptides that are involved in host sporulation and germination [21,47]. The exact role of these proteins is unknown, however in B. anthracis the lack of these genes on pXO1 is detrimental [97]. One of the more surprising findings in pBtoxis is the presence of a set of 317 genes potentially involved in the biosynthesis and export of a cyclic peptide antibiotic similar to Enterococcus faecalis AS-48 [48]. One can speculate that this peptide could provide B. thuringiensis with a competitive advantage in the environment or act as a signalling molecule. 6.1.4.2. pBT9727. B. thuringiensis 97-27 has been shown to produce crystal protein in sporulated culture by direct microscopic examination [54]. Detailed sequence analysis of pBT9727 (CP000047), the sole plasmid found in B. thuringiensis 97-27, did not reveal any genes encoding for the Cry toxin with similarity to already known B. thuringiensis toxin genes. Interestingly, pBT9727 shows similarity to B. anthracis pXO2. Comparison of the predicted coding regions of the two plasmids revealed that pBT9727 shares 89% (82/92) of its putative coding sequences with pXO2. Their replication proteins are almost identical and the predicted origin of replication is well conserved (Fig. 4, cf. [80]). The level of protein similarity, combined with the conservation of gene order, suggests that these plasmids might have diverged recently. Like pBC10987 and pXO1; pBT9727 and pXO2 share a common backbone (Fig. 3(a)). The pXO2 region encoding for the poly-c-D-glutamic acid capsule biosynthetic genes is replaced on pBT9727 with genes encoding hypothetical proteins and putative mobile elements. This replacement suggests that pBT9727 might have evolved to fulfil other functions than providing this isolate with capsule biosynthetic genes. 6.2. Replication and mobility mechanisms of the B. cereus group plasmids 6.2.1. Unidentified replicons The following plasmids have no identified replication system: pXO1, pBCXO1, pBC10987, pBC218, pBtoxis and pZK467 (Table 2). A number of hypotheses have been put forward to account for this lack of clearly identifiable replication machinery, including that these plasmids carry a novel replication mechanism [47]. Attempts have been made to identify the regions involved in the replication machinery of pXO1 using subcloning. Kaspar and Robertson [75] identified an 11 kb region that is thought to play a role in pXO1 replication. However, this region does not encode genes with similarity to any other plasmid replication system known. One gene, a type I DNA topoisomerase was identified by comparison of pBC10987 (BCEA0140, [47]) and pXO1 (BXA0213, [41]). This protein appears to be conserved in the following large B. cereus plasmids with an unidentified replication system pBc10987, pOX1 and pBCXO1, and is also found in pZK467 but where it is thought to be non-functional as it is in three gene fragments (pZK467_319-321). No homologue to this peptide is present in pBC218 or pBtoxis. In pBc10987, this type I 318 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 Fig. 4. Identification of the origin of replication structure in theta replicating plasmids of B. cereus group. (a) Alignment of the 60 nucleotide region identified by Tinsley et al. [80] as the origin of replication. The gray box highlights a conserved region containing the origin, which is identified by the arrow. Other conserved nucleotides in the region are indicated with an ‘‘*’’. The plasmids are from Bacillus anthracis pXO2 (GenBank Accession No. AE017335), B. thuringiensis pBT9727 (GenBank Accession No. AE017335), Bacillus thuringiensis pAW63 (GenBank Accession No. AJ011655) and Enterococcus faecalis pAMb1 (GenBank Accession No. AF007787). (b) The pXO2 replication region. The B. anthracis pXO2 iteron-binding region is between the copy number control gene parA and the replication gene repS. Arrows on the nucleotide sequence identify the iteron-binding repeat, ‘‘ATGTGTAA’’. There are 10 direct repeats in the forward orientation and three in the reverse orientation in pXO2. There are also other degenerate repeats in the region that are not indicated. The structure of the replication region including a similar repeated region is present in B. thuringiensis pBT9727. DNA topoisomerase may associate with a putative DNA polymerase III b-subunit thought to increase the processivity of replication [98]. Unfortunately, this second gene is not present in any of the other large B. cereus group plasmids. It may participate in pBc10987 replication but does not appear to be essential for the replication of all members of this group of plasmids. Another plasmid-borne replication gene candidate is a host-factor-like protein (BCEA0146 – pBc10987; BXA0206 – pXO1; pZK467_0115 – pZK467, ORFBT116 – pBtoxis). These peptides have nucleotide binding domains and are similar in all large B. cereus group plasmids lacking an identifiable replication system. While it is unlikely that this peptide accounts for the entire replication machinery, it is a conserved plasmid-encoded peptide. One alternate hypothesis is that B. cereus plasmids with no identifiable replication system are actually the result of reduction of an ancestral chromosome. Many of the genes found on these plasmids have significant similarity with chromosomally encoded genes from other members of the B. cereus group, B. subtilis, Bacillus halodurans and other low G+C Gram-positive species such as Listeria and Staphylococcus. Additionally, the GC skew, gene organization and gene orientation bias of these large plasmids appear more similar to that of chromosomes. In line with the chromosomal reduction theory, one can speculate that the replication machinery for these plasmids is actually chromosomally encoded and only the actual origin of replication is present on the plasmids themselves. Recent work has identified a chromosomally encoded helicase in the B. cereus group of organisms similar to that of Staphylococcus aureus plasmid pT181 [99,100]. This helicase, PcrA, functions as a nickase and can initiate replication of pT181. The B. anthracis PcrA has been shown to function as a helicase and initiate replication of pT181. It is thought that the B. cereus homologues D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 319 could perform similar function on plasmids that do not have a clearly recognizable replication system. Additionally, such a system would explain why the replication regions of these plasmids have gone undiscovered. 6.2.2. Theta replicons Based on similarity to other replication proteins identified from plasmids in the B. cereus group and other Gram-positive organisms, the origin of replication was readily identified in pXO2 [101,102]. While well characterized in B. thuringiensis plasmid pAW63, the pXO2 origin of replication was not functionally characterized until recently [80]. Tinsley et al. [80] demonstrated that the minimal replicon comprises solely of the RepS peptide (BXB0039) and the origin of replication. In addition, the functional origin of replication was shown to be limited to a 60 bp region to which RepS specifically binds (Fig. 4(a)). Furthermore, RepS was shown to bind to the single stranded form of this region, providing further evidence that RepS is responsible for the initiation of replication. pBT9727 origin of replication (59/60 nucleotides) and RepS are similar (91% amino acid identity) to that of pXO2 indicating that pBT9727 belongs to the theta replicating plasmid family. On the other hand, plasmid pZK54 is tenuously categorized as a theta replicating plasmid based on similarity of the replication associated protein, pZK54_001. pZK54 lacks the mobilization protein and the 60 nt origin of replication identified by Tinsley et al. [80], suggesting that a putative novel mechanism might be involved in pZK54 maintenance and mobility. All theta replication proteins from B. cereus group plasmids share a significant level of similarity and cluster together phylogenetically suggesting a common ancestral origin (Fig. 5(a)). 6.2.3. Rolling circle replicons All B. cereus group small plasmids (<10 kb) appear to replicate through a rolling circle mechanism. Sequence analysis and comparison of all B. cereus replication proteins, including theta and rolling circle, allow for clustering of these plasmids into distinct groups (Fig. 5). This comparison includes type sequences for each plasmid group as determined by Andrup et al. [90]. All small rolling circle plasmid replication proteins clustered together and are clearly distinct from theta replication protein sequences. pZK8 and pZK9 both clustered with group VII plasmids as defined previously [90]. pZK5 is also a group VII member but might represent an outlier to this group. Classification of the small rolling circle replicating plasmids by this sequence analysis is a robust and effective way to identify the mode of replication for such plasmids. Fig. 5. Comparison and clustering of B. cereus group plasmids based on replication and mobilization proteins. Replication or mobilization proteins, as identified by annotation, were aligned and compared using CLUSTALW. The unrooted neighbor-joining phylogenetic trees were generated amd displayed with Tree View. Theta-replicating plasmid proteins are within the blue ovals, Rolling circle replication plasmids of group IV are in the green ovals and only two members of the group III family replication proteins could be identified for inclusion. (a) Replication proteins. (b) Mobilization proteins. 6.2.4. Plasmid mobility While the B. anthracis plasmids have not been directly shown to be self-transmissible, previous reports have demonstrated that some of them can be mobilized with the help of conjugative plasmids [74]. B. thuringiensis subsp. israeliensis pXO16 is an example of such a conjugative plasmid [74,103]. Interestingly, no transfer or mobilization regions are identifiable in the sequence of B. cereus large plasmids. In contrast, the mobilization proteins encoded on the smaller B. cereus ZK and other B. thuringiensis plasmids suggests that they may be selfmobilizable but, lack the ability to create pores to transfer themselves to the recipient cells. It is possible that the combination of the mobilization capabilities of one plasmid and an unidentified membrane associated transfer system from another plasmid will allow transfer of both/all plasmids from the donor cell to a willing recipient cell. The B. cereus large plasmids harbor candidate pore formation genes, such as the TraD/G conjugation 320 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 proteins, that could form a pore through a membrane complex however their functionality has not been demonstrated experimentally. Sequencing of mobilizable B. cereus group plasmids such as pOX11 [92], pXO12 [92] or pXO16 [74,104] would advance our understanding of plasmid transfer mechanisms in this group of organisms. A number of limited studies have been undertaken that demonstrate that B. cereus group plasmid transfer is not affected by DNase, involves membrane interaction [74] and in some cases employs an Ôaggregation substanceÕ [104], all which suggest a conjugative transfer mechanism. Based on sequence analysis of the plasmids of the B. cereus group of organisms, it is evident in some cases that the significant differences observed in pathogenicity and host range are often dictated by genes carried on plasmids. As suggested for Yersina pestis [105], where pathogenicity and host range is determined by the plasmid content of an isolate, the designation of ‘‘plasmidovar’’ would be applicable to the B. cereus group of organisms, such that B. anthracis would be B. cereus plasmidovar anthracis. 7. The phage of the B. cereus group Besides plasmids, bacteriophages are another important source of gene flow in bacteria. Bacteriophages are viruses that infect bacteria. Bacteriophage can be either lytic, redirecting cellular processes for the sole purpose of producing additional virus progeny, or temperate, insetting themselves into the host bacterium genome where they are transmitted ‘‘benignly’’ from generation to generation in concert with a host replicon. Bacteriophages have an extremely high degree of host specificity and may integrate as a prophage into a unique site or multiple locations in the host genome. This specificity has been exploited as typing feature [106] and proposed as a possible therapeutic intervention in the treatment of bacterial diseases [107,108]. As an increasing number of bacterial genomes are being sequenced, more prophage are being identified and the distribution and diversity of bacteriophage is beginning to be appreciated (for recent reviews, see [109,110]). Genes carried by bacteriophage encode proteins that modulate lysogenic conversion of the host and may provide a selective advantage [111]. The prophage may remain competent to enter a lytic cycle in response to appropriate induction signals or may remain quiescent for generations. Over time, the prophage may acquire mutations that preclude their re-entry into a lytic cycle of growth, while genes originally carried by the virus can remain active in the host extending the lysogens niche over non-lysogenic isogenic strains. In fact, there are several examples of phage-derived sequences representing in excess of 10% of a sequenced bacterial genome [109]. Phage can also facilitate horizontal gene transfer and promote genomic rearrangements, a factor that has contributed to the emergence of bacterial pathogens (for review, see [112]). This is most clearly demonstrated in a comparison of the genomes of two E. coli stains, the benign K12 and the virulent O157:H7 where significant genetic differences between these two strains can be attributed to differences in prophage content [113]. While the best-characterized phage in the Grampositive Bacillus genera are from B. subtilis, similar prophages have been identified in all members of the B. cereus group. We recognize that the identification of prophage in bacterial genomes is non-trivial [109,110]. Consequently, the current estimates of the prophage contribution to the genomic content of the members of the B. cereus group should be regarded as conservative and tentative. 7.1. Genomic phage content Each of the 10 B. anthracis genome sequences contains four prophages located in the same genomic location. These prophages have been designated LambdaBa01-04 [40]. The B. anthracis prophages appear to be unique to B. anthracis, however, prophage LambdaBa01 is at least partially present in B. cereus ZK (Fig. 6). Interestingly, while the gene content appears to be conserved the Fig. 6. Blast similarities represented using Blast score ratios and visualized with TIGR MEV (multiple expression viewer). Each vertical line represents a unique gene in one of the predicted B. anthracis prophage and each horizontal column represents a different isolate. Yellow regions indicate similarity, whereas the black regions are dissimilar. This figure demonstrates that only B. cereus Zebra Killer contains one of the phage from B. anthracis. The B. anthracis phage are not generally conserved among this group and neither are the B. cereus or B. thuringiensis phage (data not shown). D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 relative genomic location is different. Whether these differences represent a subtle variation in species-specific phage integration sites, or provide an evolutionary insight into the relationship between B. cereus ZK and the B. anthracis are important questions that have not yet been resolved. Three prophages have been identified in the genome sequence B. cereus ATCC 10987, while B. cereus ATCC 14579 contains six putative integrated prophages and a linear non-integrated phage, designated pBClin15. B. cereus G9241 contains a not well-characterized cryptic phage of 29,886 bp (pBClin29, [20]) that encodes phage-like proteins and a plasmid replicon similar to B. anthracis pXO2. Interestingly, the B. thuringiensis 97-27 genome appears to contain as many as 10 prophages on the chromosome and two prophage on the pBT9727 plasmid. Including the B. anthracis LambaBa01-like prophage, B. cereus ZK genome may have as many as 27 chromosomally encoded prophages, based on the occurrence of distributed phage genes, and an additional nine in plasmid pZK467, and four in pZK54. None of the putative prophage integration sites in B. thuringiensis 97-27 or B. cereus ZK appear to be associated with tRNA genes as is often the case [114]. These high numbers of prophages may be an overestimate as some consist of only a few genes with some degree of similarity to other phages. 7.2. Lytic induction An important test of prophage competence is the ability to initiate a lytic cycle by induction. We have demonstrated the competence of several prophage to induce a lytic cycle using a standard mitomycin C treatment from a number of B. cereus and B. thuringiensis strains (Rasko, unpublished). Not surprisingly, it was shown that the host-range of these phages is restricted to the species or strain from which they were obtained. However, there is one notable exception, strain B. cereus ATCC 4342 can be used to propagate the Gamma and Cherry phages that were previously thought to be limited to B. anthracis strains [106,115]. The factors responsible for the expansion of the phage range are currently under investigation. Finally, it is interesting to note that typing with MLST places B. cereus ATCC 4342 in the phylogenetic clade that contains B. anthracis and may represent a close evolutionary relationship (Fig. 1). 7.3. Exploitation of phage Given the high degree of specificity and limited host range of bacteriophage, it seems quite reasonable to use them and their encoded activities as a means of strain identification and infection control. 321 7.3.1. Use as a typing tool for B. anthracis (Gamma and Cherry phage) While there are four prophages in each of the B. anthracis genomes sequenced to date, clinical laboratories utilize, in combination with a number of biochemical tests, cell lysis with Gamma phage as a phenotypic characteristic in the identification of B. anthracis [106]. Sensitivity to Gamma phage remains highly specific to B. anthracis, however B. cereus ATCC 4342 is also sensitive to Gamma phage. As more isolates closely related to B. anthracis are being isolated with pathogenic characteristics, Gamma phage sensitivity among B. cereus isolates will become increasingly common and will reduce the discriminating power of this assay to differentiate B. anthracis from B. cereus. 7.3.2. Use of phage lysis protein as a biological control While the complete sequence for Gamma phage is not yet available, its lysis protein, PlyG, has been cloned, sequenced and characterized [115]. The lytic activity appears to be restricted to B. anthracis, however the specificity of this activity has not been fully elucidated. It has been suggested that the lysin proteins from the lysogenic phages of B. anthracis could be utilized as a method for biological control. The specificity for B. anthracis of such proteins has been previously demonstrated [115] and used for clinical typing of B. anthracis (see above). This activity may also be applied to specifically lyse B. anthracis in other situations, such as treatment of infections. The possibility of using these proteins as biological control mechanisms holds promise, but cross reactivity with certain B. cereus strains could be a problem [115] and, more importantly, the lytic activity of these proteins is limited to vegetative cells. These proteins do not affect B. anthracis spores, the infectious particles. A strategy has been suggested that employs the use of spore germinant solutions to induce the outgrowth of vegetative forms in combination with a lysin solution to destroy emergent vegetative cells. The rapid concerted conversion from spores to vegetative forms (less than 10 min) [116] suggests that this approach could be applied successfully. Both the germinant and lysin solution should be harmless enough to both man and machine to use not only in the case of a bioterror attack, but also as prophylaxis for the treatment of troops and machines returning from anthrax contaminated areas. Additionally, since the phage lysin proteins are thought to attack the basic building blocks of the cell wall, it would be unlikely that resistance to these methods will develop as rapidly as antibiotic resistance. Interestingly, no equivalent system using phages has been proposed or examined for B. cereus or B. thuringiensis, certainly due to the fact that a phage would be specific to one strain but not for the entire group. 322 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 8. Functional genomics of the B. cereus group of organisms Since the publication of the first B. cereus group organism genomes [40,45], a significant number of studies have taken advantage of the sequence to generate functional data. Not surprisingly B. anthracis has been the focus of much of this work. The mail anthrax attacks of 2001 provided an additional impetus for research as well as an infusion of funding for the development of microarray and proteomic research programs. 8.1. Transcriptional analysis using microarrays 8.1.1. B. cereus/B. thuringiensis The availability of B. cereus specific arrays by a limited number of companies (NimbleGen and Qiagen) has not yet generated any published data. Additionally, a commercially available B. thuringiensis specific array has not been advertised. While these species-specific arrays are useful, most information would derive from an array that contains the B. cereus core genomic elements as well as the unique genes from a number of species or isolates. The advantage of building such a chimeric array would be to capture the level of conservation of any interrogated isolate in relation to the core genotype, but it would also detect the level of divergence and possible gene transfer between isolates/species in comparative genomic hybridization (CGH) experiments. In addition to obtaining a metric for the level of conservation, the chimeric array will allow representation of multiple strains, isolates or species on a single array without the need for redundant coding sequences to be represented, thus saving space, time, effort and money. 8.1.2. B. anthracis A B. anthracis Ames microarray (both 70-mer oligonucleotide and amplicon-based) is available through the NIAID funded Pathogen Functional Genomics Resource Center (http://pfgrc.tigr.org/). Other arrays have been produced and used for species typing [117] and resequencing [118]. Read et al. [40] utilized an amplicon-based array as a comparative CGH tool to interrogate the genomic content of 19 diverse B. cereus group organisms. This diverse set included a number of the B. cereus strains from clinical sources, mostly periodontal infections from Norway [3,28,62] as well as strains from environmental sources and common laboratory strains including B. cereus ATCC 14579 and B. cereus ATCC 10987 [45,47]. This study revealed a high degree of genomic similarity among these 19 B. cereus isolates. In addition, it indicates the presence of pXO1 gene homologues in half of the 19 strains examined, consistent with other studies [63]. In contrast, very few homologues of pXO2 genes were found to hybridize [40]. B. cereus isolates from clinical sources contained more pXO1 and/or pXO2 genes than non-clinical isolates, suggesting a pathogenic role for plasmid-encoded genes. While this technique does not allow the determination of gene order, it confirms the presence of plasmid-like genes in these species. Molecular examination of some of these clinical isolates has revealed the presence of large plasmids (>200 kb) that are currently under investigation (Rasko, unpublished data). Using the ratios obtained for chromosomal genes, it was possible to reconstruct the phylogenetic relationship of these 19 isolates – the clustering obtained was consistent with that obtained using other methods [3,8,27– 29,32,33]. In addition to CGH experiments, this array has also used for expression analysis in two separate studies. The expression of plasmid-encoded virulence factors and genes that are involved with their regulation were analyzed. Comparison of expression patterns for wild type and B. anthracis AtxA knockout mutants demonstrated that AtxA is the dominant regulatory protein and appears to be a master virulence regulator in B. anthracis [71]. AtxA was shown to regulate AcpA and AcpB, which were previously thought to regulate pXO2encoded capsule biosynthesis. B. anthracis gene expression patterns were analyzed as the bacterium progressed through logarithmic phase and entered sporulation [72]. In contrast to the previous study, Liu et al. did not make use of mutational analysis to examine the regulation but rather took a less invasive method to examine gene expression as growth progressed to unravel the regulatory cascade associated with spore formation. Five distinct waves of gene expression containing 36% of all predicted B. anthracis genes were observed. The five waves roughly corresponded to early, mid or late logarithmic growth, stationary phase growth and spore formation. The data revealed that the sporulation program in B. anthracis is similar to the well-characterized B. subtilis system [119]. However, B. anthracis sigma factor A (SigA) expression patterns deviate from those of its B. subtilis counterpart. sigA is induced during the final stages of sporulation in B. anthracis whereas in B. subtilis SigA is produced during vegetative growth [119]. Additional examination of the expression data demonstrated that the plasmid-encoded virulence factors were expressed only in early logarithmic phase. The true power of this study is that the expression data were combined with a proteomic analysis of the sporulating cultures [72]. 8.2. Proteomics studies 8.2.1. B. anthracis The genome sequence and its predicted proteome have allowed for high-throughput proteomic analyses D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 where proteins are enzymatically digested and the resulting peptides analyzed with two-dimensional liquid chromatography coupled with tandem mass spectrometry analysis. Peptide molecular weights are then matched bioinformatically to specific proteins. Combining this approach with expression analysis in a single study highlights the complementary nature of genomic and proteomic analyses. Liu et al. [72] applied such an approach to the study of spore formation and were able to rapidly document high-resolution temporal changes in gene expression associated with spore formation, while proteomic analysis provided a detailed snapshot of the protein content and relative abundance in the spore. Surprisingly, genes that were upregulated in the final stages of spore formation were rarely identified by the proteomic analysis as being constituents of the spore. Of the 873 genes identified to be upregulated in the final stages preceding sporulation, only 173 were identified proteomically as being spore components [72]. This suggests that the sporulating cells obtain proteins from pre-existing peptide pools to form the spore. If the microarray and proteomic data had not been combined into a single study, a number of incorrect conclusions as to the temporal origin of the proteins in the spore could have been made. While the study by Liu et al. [72] demonstrated the power of using proteomic analysis in a high-throughput mode, other proteomic studies are focusing on rapid and accurate speciation of B. anthracis or B. cereus based on the proteomic content of their spore employing a combined 2D-gel electrophoresis and tandem mass spectrometry approach [120]. These studies have been successful in differentiating spores and vegetative forms of B. anthracis from its close relatives [120,121]. In addition to examining and differentiating the Bacillus species, these proteomic studies have identified potential new spore coat proteins not previously annotated as such or known to reside in the membrane or spore surface [122]. Anthrax vaccine preparations have also been characterized through proteomic analysis. A recent study using 2D gel-electrophoresis [123] confirmed that the major constituent of the B. anthracis vaccine was the protective antigen, but identified a number of minor contaminating products. These minor constituents included proteolytic cleavage products of the protective antigen, lethal factor and edema factor as well as other putative virulence factors such as EA1, Sap and S-layer proteins. Additionally, cell constituents could be identified such as 60 kDa heat-shock protein, enolase, fructose-bisphosphate aldolase and nucleoside diphosphate kinase [123]. Proteomics can also be applied to examine the spore or bacterial surface for novel vaccine candidates. 2D gel electrophoresis of the spore proteome probed with sera from immunized animals identified eight in vivo immunogens, six of which were previously reported as anti- 323 genic targets. Five of the eight candidates were preselected through bioinformatic analysis of the genome sequence of B. anthracis [124]. This study highlights that a combination of genomic data, bioinformatics analysis and proteomics can contribute to novel anthrax vaccine development. 8.2.2. B. cereus Unlike B. anthracis, proteomic analyses were applied to B. cereus vegetative cells and not spores. B. cereus PlcR mutant strains were compared to the wild type using protein 2D gel electrophoresis [54]. As expected, the inactivation of plcR in most cases abolished or significantly reduced the expression of PlcR-regulated peptides. These peptides included a number of virulence factors known to be under the control of PlcR, such as collagenase, hemolysins, proteases and toxins. Those directly regulated by PlcR were most significantly affected, whereas those indirectly regulated were reduced in expression levels. Detailed 2D gel electrophoresis proteomic analysis of B. cereus biofilm establishment and maintenance on glass wool has been reported [125,126]. In the dairy industry, B. cereus biofilms inside storage tanks and associated piping can create problems for product sterility and longevity. Using a proteomic approach, B. cereus isolates in biofilms have been shown to express at least 10 additional proteins and lack seven proteins during adhesion to a solid surface and establishment of the biofilm [125]. These peptides may play important roles in the maintenance of the biofilm and/ or represent metabolic changes triggered when the bacterium switches from a planktonic to a sessile lifestyle. 8.2.3. B. thuringiensis While there are few published B. thuringiensis proteomic studies, significant progress have been made in the identification of sporulation regulatory pathways using MALDI-TOF (matrix-assisted laser desorption/ionization time of flight) analysis [127]. These pathways are a driver of B. thuringiensis biology, as the spore is the bacterial form used as a biopesticide. Other proteomic studies have focused on the identification of the Cry toxin receptor in the Manduca sexta midgut using 1D and 2D gel electrophoresis [128]. These different steps are essential in the life cycle of B. thuringiensis – proteomics could have a major impact in developing a better understanding of bacterial virulence mechanisms, both by studying the host and the bacterium itself. Overall, proteomics and microarray studies have opened the opportunity to leverage the genomic information of the members of this group. These studies have enabled a better understanding of spore formation regulatory cascades, identified unknown regulatory elements and pathways, and have started to examine the 324 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 interactions of the B. cereus group of organisms with their respective environments. 9. Summary: One species on the basis of genomic evidence? Attempts at bacterial systematics began long before the discovery of DNA as the hereditable material. In fact, the seminal discovery demonstrating that DNA was the genetic material, through the transformation of avirulent pneumococci to the virulent form [129] hinted at the importance of horizontal gene transfer as a mechanism to increase genetic diversity and thereby niche expansion by a microbial species. Bacteria were originally classified largely on the basis of phenotype, morphology, ecology and/or associated disease state. For example, the bacteria that are the subject of this review were named Bacillus for their rod-shape, cereus for presumably an association with cereal crops, and anthracis as the cause of the disease anthrax. The criteria for the ÔspeciesÕ designation have been widely debated, however the tenet of reproductive isolation is largely unassailable. This principle seemed inviolate in asexually reproducing bacteria until the demonstration of ‘‘fertility’’ in Escherichia coli in the early 1950s [130,131]. The subsequent discovery that antibiotic resistance could be disseminated between different species within the Enterobacteriacae via plasmids [132] caused reconsideration of the basis of bacterial classification. Classical bacterial systematics is now being challenged by discoveries being promulgated by the genomics revolution. The defining characteristics of a species must be grounded in its genetic/genomic architecture. In the B. cereus group of organisms, virulence and pathogenicity appear to be promiscuous and spread with plasmids. As reviewed in this paper, it is clear that the bacterial chromosomes of the sequenced members of this group are extremely similar. These chromosomes show a high level of synteny and protein identity, a combination never observed between different bacterial species. Furthermore, there is evidence for a shared set of core putative virulence factors between different pathogenic and non-pathogenic members of the group. Very few chromosomal genes or sets of genes are unique to one species. These regions often represent metabolic adaptations, and in many cases are found in another species with different phenotypic and pathogenic traits. Their presence cannot be associated with a specific subset of organisms. Conversely, much of the disease and host specificity in this group can be attributed to plasmid content (i.e., pXO1 and pXO2 in B. anthracis). The pXO1-like plasmid in B. cereus G9241 has been hypothesized to be responsible for the anthrax-like clinical presentation caused by this isolate. However, in other isolates no role can be associated with any of the harbored plasmids. Interestingly, clustering of these plasmids based on the similarity of their replication systems is possible, offering an alternative method of systematic classification. Certainly our analyses demonstrate a bias toward significant pathogens of the group. However, not all members of the group are highly virulent. While 15 genome sequences are publicly available for a diverse set of B. cereus group organisms, genome data analysis has not delivered in developing a better understanding of the mechanisms that contribute to, and limit the acquisition of virulence. Access to such information will help to further refine the definition of a species. Technological advances in functional genomics and proteomics are giving scientists the opportunity to leverage genome sequence data. From the analysis presented here and those of others, it is apparent that gene content might play a decreased role in the diversity of phenotype and pathogenicity observed in the B. cereus group. Subtle changes to regulatory networks may be responsible for the range of phenotypic traits displayed by the B. cereus group members. Functional studies and genetic linkage experiments, combined with proteomic analysis should provide a better understanding of these regulatory pathways and their genetic basis. In addition, using these technologies combined with access to the genomes of the human, pathogenic and non-pathogenic members of the group, it is now possible to study the interaction of the bacterium with its host. This may allow the emergence of a detailed molecular understanding of the pathogenicity of this group of organisms that will benefit the development of treatment and prevention measures. The evolution of B. anthracis as a highly successful pathogen causing a readily identifiable disease has led to its over-representation in culture collections of isolates from the B. cereus group. Thus, should B. anthracis be considered an oversampled B. cereus? Based on genomic analyses of representatives of the group, these isolates carry different plasmids in a similar genetic background. Only subtle differences in gene content and protein similarities are observed when the chromosomes of members of the group are compared. While it is true that B. anthracis can be readily differentiated from B. cereus based on biochemical tests [33], a ÔproblemÕ still exists for borderline isolates such as the pathogenic B. cereus G9241 [20], where these tests fail to recognize the pathogenic potential of this isolate. The limited definition of the nature of B. anthracis emphasizes the challenges faced by public health officials when confronted with non-B. anthracis pathogenic isolates, and their inability to identify them as such [20]. The genome of B. cereus G9241 would be indistinguishable from these of any other B. cereus isolates, if it did not contain a homologue of B. anthracis pXO1. Classification of the D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 members of the B. cereus group of organisms has long been the source of controversy. In 1952, based on the observation that an isolate of B. anthracis had lost both virulence, and its plasmids, and was indistinguishable from B. cereus in term of pathogenicity, Smith et al. [133] concluded ‘‘B. anthracis is taxonomically a pathogenic variety of B. cereus’’. The bacterium was later listed as B. cereus var anthracis [134], a nomenclature questioned ever since [37,38]. While genome sequence analysis has not elucidated the pathogenic potential of some isolates of the group, it has offered unprecedented insights into the core of their making. Based on the chromosome genomic comparison reviewed in this paper, it is not possible to distinguish members of the B. cereus group from one another. Should they be considered one species based on genomic evidence? It is recognized that for economical and social reasons, changing the current nomenclature would be quite a challenge. It is hoped that this paper may represent a starting point for discussion and further novel studies through focusing on the similarities and distinctions that contribute to the nature of this group of bacteria. Acknowledgments D.A.R. and J.R. are supported with Federal funds from the National Institute of Allergy and Infectious Disease, National Institutes of Health, under Contract No. N01-AI15447. M.R.A. and C.S.H. are supported by the US Department of Energy Contract W-7405ENG-36 and LAUR#04-8482. We are grateful to Garry Myers, Thomas Brettin and Paul Jackson for their comments during the preparation of this manuscript and to Gary Xie for his technical assistance. References [1] Chen, M.L. and Tsen, H.Y. (2002) Discrimination of Bacillus cereus and Bacillus thuringiensis with 16S rRNA and gyrB gene based PCR primers and sequencing of their annealing sites. Journal of Appl. Microbio. 92, 912– 919. [2] Daffonchio, D., Cherif, A. and Borin, S. (2000) Homoduplex and heteroduplex polymorphisms of the amplified ribosomal 16S–23S internal transcribed spacers describe genetic relationships in the ‘‘Bacillus cereus group’’. Applied and Environmental Microbiology 66, 5460–5468. [3] Helgason, E., Økstad, O.A., Caugant, D.A., Johansen, H.A., Fouet, A., Mock, M., Hegna, I. and Kolstø, A.B. (2000) Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis – One species on the basis of genetic evidence. Applied and Environmental Microbiology 66, 2627–2630. [4] Lecadet, M.M., Frachon, E., Dumanoir, V.C., Ripouteau, H., Hamon, S., Laurent, P. and Thiery, I. (1999) Updating the Hantigen classification of Bacillus thuringiensis. Journal of Applied Microbiology 86, 660–672. 325 [5] Orduz, S., Restrepo, W., Patino, M.M. and Rojas, W. (1995) Transfer of toxin genes to alternate bacterial hosts for mosquito control. Memorias Do Instituto Oswaldo Cruz 90, 97–107. [6] Schnepf, E., Crickmore, N., Van Rie, J., Lereclus, D., Baum, J., Feitelson, J., Zeigler, D.R. and Dean, D.H. (1998) Bacillus thuringiensis and its pesticidal crystal proteins. Microbiology and Molecular Biology Reviews 62, 775–790. [7] Jensen, G.B., Hansen, B.M., Eilenberg, J. and Mahillon, J. (2003) The hidden lifestyles of Bacillus cereus and relatives. Environmental Microbiology 5, 631–640. [8] Helgason, E., Caugant, D.A., Lecadet, M.M., Chen, Y.H., Mahillon, J., Lovgren, A., Hegna, I., Kvaloy, K. and Kolstø, A.B. (1998) Genetic diversity of Bacillus cereus and B. thuringiensis isolates from natural sources. Current Microbiology 37, 80–87. [9] Hernandez, E., Ramisse, F., Cruel, T., le Vagueresse, R. and Cavallo, J.D. (1999) Bacillus thuringiensis serotype H34 isolated from human and insecticidal strains serotypes 3a3b and H14 can lead to death of immunocompetent mice after pulmonary infection. FEMS Immunology Medical Microbiology 24, 43–47. [10] Hernandez, E., Ramisse, F., Ducoureau, J.P., Cruel, T. and Cavallo, J.D. (1998) Bacillus thuringiensis subsp. konkukian (serotype H34) superinfection: case report and experimental evidence of pathogenicity in immunosuppressed mice. Journal of Clinical Microbiology 36, 2138–2139. [11] Hernandez, E., Ramisse, F., Gros, P. and Cavallo, J. (2000) Superinfection by Bacillus thuringiensis H34 or 3a3b can lead to death in mice infected with the influenza A virus. FEMS Immunology Medical Microbiology 29, 177–181. [12] Logan, N.A. and Turnbull, P.C. (1999) (Murray, P.R., Ed.), Manual of Clinical Microbiology, pp. 357–369. American Society for Microbiology, Washington, DC. [13] Drobniewski, F.A. (1993) Bacillus cereus and related species. Clinical Microbiology Reviews 6, 324–338. [14] Margulis, L., Jorgensen, J.Z., Dolan, S., Kolchinsky, R., Rainey, F.A. and Lo, S.C. (1998) The arthromitus stage of Bacillus cereus: intestinal symbionts of animals. Proceedings of the National Academy of Sciences of the United States of America 95, 1236–1241. [15] A.B. Kolstø, D. Lereclus, M. Mock, Genome structure and evolution of the Bacillus cereus group, in: Pathogenicity Islands and the Evolution of Pathogenic Microbes, vol. 2, 2002, pp. 95– 108. [16] Jernigan, D.B., Raghunathan, P.L., Bell, B.P., Brechner, R., Bresnitz, E.A., Butler, J.C., Cetron, M., Cohen, M., Doyle, T., Fischer, M., Greene, C., Griffith, K.S., Guarner, J., Hadler, J.L., Hayslett, J.A., Meyer, R., Petersen, L.R., Phillips, M., Pinner, R., Popovic, T., Quinn, C.P., Reefhuis, J., Reissman, D., Rosenstein, N., Schuchat, A., Shieh, W.J., Siegal, L., Swerdlow, D.L., Tenover, F.C., Traeger, M., Ward, J.W., Weisfuse, I., Wiersma, S., Yeskey, K., Zaki, S., Ashford, D.A., Perkins, B.A., Ostroff, S., Hughes, J., Fleming, D., Koplan, J.P. and Gerberding, J.L. (2002) Investigation of bioterrorism-related anthrax, United States, 2001. Epidemiologic Findings 8, 1019–1028. [17] Jernigan, J.A., Stephens, D.S., Ashford, D.A., Omenaca, C., Topiel, M.S., Galbraith, M., Tapper, M., Fisk, T.L., Zaki, S., Popovic, T., Meyer, R.F., Quinn, C.P., Harper, S.A., Fridkin, S.K., Sejvar, J.J., Shepard, C.W., McConnell, M., Guarner, J., Shieh, W.J., Malecki, J.M., Gerberding, J.L., Hughes, J.M. and Perkins, B.A. (2001) Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerging Infectious Diseases 7, 933–944. [18] Turnbull, P.C.B. (2002) Introduction: anthrax history, disease and ecology. Anthrax 271, 1–19. [19] Mock, M. and Fouet, A. (2001) Anthrax. Annual Review of Microbiology 55, 647–671. 326 D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 [20] Hoffmaster, A.R., Ravel, J., Rasko, D.A., Chapman, G.D., Chute, M.D., Marston, C.K., De, B.K., Sacchi, C.T., Fitzgerald, C., Mayer, L.W., Maiden, M.C., Priest, F.G., Barker, M., Jiang, L., Cer, R.Z., Rilstone, J., Peterson, S.N., Weyant, R.S., Galloway, D.R., Read, T.D., Popovic, T. and Fraser, C.M. (2004) Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proceedings of the National Academy of Sciences of the United States of America 101, 8449–8454. [21] Okinaka, R., Cloud, K., Hampton, O., Hoffmaster, A., Hill, K., Keim, P., Koehler, T., Lamke, G., Kumano, S., Manter, D., Martinez, Y., Ricke, D., Svensson, R. and Jackson, P. (1999) Sequence, assembly and analysis of pXO1 and pXO2. Journal of Applied Microbiology 87, 261–262. [22] Okinaka, R.T., Cloud, K., Hampton, O., Hoffmaster, A.R., Hill, K.K., Keim, P., Koehler, T.M., Lamke, G., Kumano, S., Mahillon, J., Manter, D., Martinez, Y., Ricke, D., Svensson, R. and Jackson, P.J. (1999) Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. Journal of Bacteriology 181, 6509–6515. [23] Priest, F.G. (1981) DNA homology in the genus Bacillus In: The Aerobic Endospore-forming Bacteria (Berkeley, R.C.W. and Goodfellow, M., Eds.), pp. 35–57. Academic Press, London. [24] Ash, C. and Collins, M.D. (1992) Comparative analysis of 23S ribosomal RNA gene sequences of Bacillus anthracis and emetic Bacillus cereus determined by PCR direct sequencing. FEMS Microbiology Letters 94, 75–80. [25] Ash, C., Farrow, J.A.E., Dorsch, M., Stackebrandt, E. and Collins, M.D. (1991) Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S ribosomal RNA. International Journal of Systematic Bacteriology 41, 343–346. [26] Bavykin, S.G., Lysov, Y.P., Zakhariev, V., Kelly, J.J., Jackman, J., Stahl, D.A. and Cherni, A. (2004) Use of 16S rRNA, 23S rRNA, and gyrB gene sequence analysis to determine phylogenetic relationships of Bacillus cereus group microorganisms. Journal of Clinical Microbiology 42, 3711–3730. [27] Carlson, C.R., Caugant, D.A. and Kolstø, A.B. (1994) Genotypic diversity among Bacillus cereus and Bacillus thuringiensis strains. Applied and Environmental Microbiology 60, 1719– 1725. [28] Helgason, E., Tourasse, N.J., Meisal, R., Caugant, D.A. and Kolstø, A.B. (2004) Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Applied and Environmental Microbiology 70, 191–201. [29] Priest, F.G., Barker, M., Baillie, L.W., Holmes, E.C. and Maiden, M.C. (2004) Population structure and evolution of the Bacillus cereus group. Journal of Bacteriology 186, 7959–7970. [30] Ko, K.S., Kim, J.W., Kim, J.M., Kim, W., Chung, S.I., Kim, I.J. and Kook, Y.H. (2004) Population structure of the Bacillus cereus group as determined by sequence analysis of six housekeeping genes and the plcR gene. Infection and Immunity 72, 5253–5261. [31] Hill, K.K., Ticknor, L.O., Okinaka, R.T., Asay, M., Blair, H., Bliss, K.A., Laker, M., Pardington, P.E., Richardson, A.P., Tonks, M., Beecher, D.J., Kemp, J.D., Kolstø, A.B., Wong, A.C.L., Keim, P. and Jackson, P.J. (2004) Fluorescent amplified fragment length polymorphism analysis of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis isolates. Applied and Environmental Microbiology 70, 1068–1080. [32] Keim, P., Kalif, A., Schupp, J., Hill, K., Travis, S.E., Richmond, K., Adair, D.M., Hugh-Jones, M., Kuske, C.R. and Jackson, P. (1997) Molecular evolution and diversity in Bacillus anthracis as detected by amplified fragment length polymorphism markers. Journal of Bacteriology 179, 818–824. [33] Ticknor, L.O., Kolstø, A.B., Hill, K.K., Keim, P., Laker, M.T., Tonks, M. and Jackson, P.J. (2001) Fluorescent amplified [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] fragment length polymorphism analysis of Norwegian Bacillus cereus and Bacillus thuringiensis soil isolates. Applied and Environmental Microbiology 67, 4863–4873. Cherif, A., Brusetti, L., Borin, S., Rizzi, A., Boudabous, A., Khyami-Horani, H. and Daffonchio, D. (2003) Genetic relationship in the ÔBacillus cereus groupÕ by rep-PCR fingerprinting and sequencing of a Bacillus anthracis-specific rep-PCR fragment. Journal of Applied Microbiology 94, 1108–1119. Radnedge, L., Agron, P.G., Hill, K.K., Jackson, P.J., Ticknor, L.O., Keim, P. and Andersen, G.L. (2003) Genome differences that distinguish Bacillus anthracis from Bacillus cereus and Bacillus thuringiensis. Applied and Environmental Microbiology 69, 2755–2764. Keim, P., Price, L.B., Klevytska, A.M., Smith, K.L., Schupp, J.M., Okinaka, R., Jackson, P.J. and Hugh-Jones, M.E. (2000) Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. Journal of Bacteriology 182, 2928–2936. Turnbull, P.C., Hutson, R.A., Ward, M.J., Jones, M.N., QUinn, C.P., Finnie, N.J., Duggleby, C.J., Kramer, J.M. and Melling, J. (1992) Bacillus anthracis but not always anthrax. Journal of Applied Bacteriology 72, 21–28. Turnbull, P.C. (1999) Definitive identification of Bacillus anthracis – a review. Journal of Applied Microbiology 87, 237–240. Fleischmann, R.D., Adams, M.D., White, O., Clayton, R.A., Kirkness, E.F., Kerlavage, A.R., Bult, C.J., Tomb, J.F., Dougherty, B.A. and Merrick, J.M. (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–512. Read, T.D., Peterson, S.N., Tourasse, N., Baillie, L.W., Paulsen, I.T., Nelson, K.E., Tettelin, H., Fouts, D.E., Eisen, J.A., Gill, S.R., Holtzapple, E.K., Økstad, O.A., Helgason, E., Rilstone, J., Wu, M., Kolonay, J.F., Beanan, M.J., Dodson, R.J., Brinkac, L.M., Gwinn, M., DeBoy, R.T., Madpu, R., Daugherty, S.C., Durkin, A.S., Haft, D.H., Nelson, W.C., Peterson, J.D., Pop, M., Khouri, H.M., Radune, D., Benton, J.L., Mahamoud, Y., Jiang, L.X., Hance, I.R., Weidman, J.F., Berry, K.J., Plaut, R.D., Wolf, A.M., Watkins, K.L., Nierman, W.C., Hazen, A., Cline, R., Redmond, C., Thwaite, J.E., White, O., Salzberg, S.L., Thomason, B., Friedlander, A.M., Koehler, T.M., Hanna, P.C., Kolstø, A.B. and Fraser, C.M. (2003) The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 423, 81–86. Read, T.D., Salzberg, S.L., Pop, M., Shumway, M., Umayam, L., Jiang, L.X., Holtzapple, E., Busch, J.D., Smith, K.L., Schupp, J.M., Solomon, D., Keim, P. and Fraser, C.M. (2002) Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 296, 2028–2033. Pearson, T., Busch, J.D., Ravel, J., Read, T.D., Rhoton, S.D., UÕRen, J.M., Simonson, T.S., Kachur, S.M., Leadem, R.R., Cardon, M.L., Van Ert, M.N., Huynh, L.Y., Fraser, C.M. and Keim, P. (2004) Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proceedings of the National Academy of Sciences of the United States of America 101, 13536–13541. Baillie, L. and Read, T.D. (2001) Bacillus anthracis, a bug with attitude!. Current Opinions in Microbiology 4, 78–81. Turnbull, P.C. (1991) Anthrax vaccines: past, present and future. Vaccine 9, 533–539. Ivanova, N., Sorokin, A., Anderson, I., Galleron, N., Candelon, B., Kapatral, V., Bhattacharyya, A., Reznik, G., Mikhailova, N., Lapidus, A., Chu, L., Mazur, M., Goltsman, E., Larsen, N., DÕSouza, M., Walunas, T., Grechkin, Y., Pusch, G., Haselkorn, R., Fonstein, M., Ehrlich, S.D., Overbeek, R. and Kyrpides, N. (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423, 87–91. D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 [46] Sneath, P.H.A. (1986) Endospore-forming Gram-positive rods and cocci (Sneath, P.H.A., Mair, N.S., Sharpe, M.E. and Holt, J.G., Eds.), BergeyÕs Manual of Systematic Bacteriology, vol. 2, p. 1131. Williams & Wilkins, Blatimore. [47] Rasko, D.A., Ravel, J., Økstad, O.A., Helgason, E., Cer, R.Z., Jiang, L., Shores, K.A., Fouts, D.E., Tourasse, N.J., Angiuoli, S.V., Kolonay, J., Nelson, W.C., Kolstø, A.B., Fraser, C.M. and Read, T.D. (2004) The genome sequence of Bacillus cereus ATCC 10987 reveals metabolic adaptations and a large plasmid related to Bacillus anthracis pXO1. Nucleic Acids Research 32, 977–988. [48] Berry, C., OÕNeil, S., Ben-Dov, E., Jones, A.F., Murphy, L., Quail, M.A., Holden, M.T.G., Harris, D., Zaritsky, A. and Parkhill, J. (2002) Complete sequence and organization of pBtoxis, the toxin-coding plasmid of Bacillus thuringiensis subsp israelensis. Applied and Environmental Microbiology 68, 5082– 5095. [49] Cummings, C.A. and Relman, D.A. (2002) Genomics and microbiology. Microbial forensics – ‘‘cross-examining pathogens’’. Science 296, 1976–1979. [50] CDC, Update: investigation of anthrax associated with intentional exposure and interim public health guidelines, October, Morbidity and Mortality Weekly Reports 50 (2001) 889–893. [51] Keim, P., Van Ert, M.N., Pearson, T., Vogler, A.J., Huynh, L.Y. and Wagner, D.M. (2004) Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infection, Genetics and Evolution 4, 205–213. [52] Suyama, M. and Bork, P. (2001) Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends in Genetics 17, 10–13. [53] Parkhill, J. and Berry, C. (2003) Genomics: relative pathogenic values. Nature 423, 23–25. [54] Gohar, M., Økstad, O.A., Gilois, N., Sanchis, V., Kolstø, A.B. and Lereclus, D. (2002) Two-dimensional electrophoresis analysis of the extracellular proteome of Bacillus cereus reveals the importance of the PlcR regulon. Proteomics 2, 784–791. [55] Lereclus, D., Agaisse, H., Gominet, M., Salamitou, S. and Sanchis, V. (1996) Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. Journal of Bacteriology 178, 2749–2756. [56] Slamti, L. and Lereclus, D. (2002) A cell–cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO Journal 21, 4550–4559. [57] Slamti, L., Perchat, S., Gominet, M., Vilas-Boas, G., Fouet, A., Mock, M., Sanchis, V., Chaufaux, J., Gohar, M. and Lereclus, D. (2004) Distinct mutations in PlcR explain why some strains of the Bacillus cereus group are nonhemolytic. Journal of Bacteriology 186, 3531–3538. [58] Mignot, T., Mock, M., Robichon, D., Landier, A., Lereclus, D. and Fouet, A. (2001) The incompatibility between the PlcR- and AtxA-controlled regulons may have selected a nonsense mutation in Bacillus anthracis. Molecular Microbiology 42, 1189– 1198. [59] Pomerantsev, A.P., Pomerantseva, O.M. and Leppla, S.H. (2004) A spontaneous translational fusion of Bacillus cereus PlcR and PapR activates transcription of PlcR-dependent genes in Bacillus anthracis via binding with a specific palindromic sequence. Infection and Immunity 72, 5814–5823. [60] Økstad, O.A., Gominet, M., Purnelle, B., Rose, M., Lereclus, D. and Kolstø, A.B. (1999) Sequence analysis of three Bacillus cereus loci carrying PIcR-regulated genes encoding degradative enzymes and enterotoxin. Microbiology 145, 3129–3138. [61] Beecher, D.J., Schoeni, J.L. and Wong, A.C.L. (1995) Enterotoxic activity of hemolysin BL from Bacillus cereus. Infection and Immunity 63, 4423–4428. 327 [62] Helgason, E., Caugant, D.A., Olsen, I. and Kolstø, A.B. (2000) Genetic structure of population of Bacillus cereus and Bacillus thuringiensis isolates associated with periodontitis and other human infections. Journal of Clinical Microbiology 38, 1615– 1622. [63] Pannucci, J., Okinaka, R.T., Williams, E., Sabin, R., Ticknor, L.O. and Kuske, C.R. (2002) DNA sequence conservation between the Bacillus anthracis pXO2 plasmid and genomic sequence from closely related bacteria. BMC Genomics 3, 34. [64] Gladstone, G.P. (1946) Immunity to anthrax – protective antigen present in cell-free culture filtrates. British Journal of Experimental Pathology 27, 394–418. [65] Lacy, D.B. and Collier, R.J. (2002) Structure and function of anthrax toxin. Anthrax 271, 61–85. [66] Friedlander, A.M. (1990) The anthrax toxins In: Trafficking of Bacterial Toxins (Saelinger, C.B., Ed.), pp. 121–138. CRC Press, Boca Raton, FL. [67] Leppla, S.H. (1995) Anthrax toxins In: Bacterial Toxins and Virulence Factors in Disease (Moss, J., Iglewski, B., Vaughn, M. and Tu, A.T., Eds.), pp. 543–572. Dekker, New York, NY. [68] Mogridge, J., Mourez, M. and Collier, R.J. (2001) Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. Journal of Bacteriology 183, 2111–2116. [69] Pannifer, A.D., Wong, T.Y., Schwarzenbacher, R., Renatus, M., Petosa, C., Bienkowska, J., Lacy, D.B., Collier, R.J., Park, S., Leppla, S.H., Hanna, P. and Liddington, R.C. (2001) Crystal structure of the anthrax lethal factor. Nature 414, 229–233. [70] Singh, Y., Khanna, H., Chopra, A.P. and Mehra, V. (2001) A dominant negative mutant of Bacillus anthracis protective antigen inhibits anthrax toxin action in vivo. Journal of Biological Chemistry 276, 22090–22094. [71] Bourgogne, A., Drysdale, M., Hilsenbeck, S.G., Peterson, S.N. and Koehler, T.M. (2003) Global effects of virulence gene regulators in a Bacillus anthracis strain with both virulence plasmids. Infection and Immunity 71, 2736–2743. [72] Liu, H., Bergman, N.H., Thomason, B., Shallom, S., Hazen, A., Crossno, J., Rasko, D.A., Ravel, J., Read, T.D., Peterson, S.N., Yates 3rd, J. and Hanna, P.C. (2004) Formation and composition of the Bacillus anthracis endospore. Journal of Bacteriology 186, 164–178. [73] Coker, P.R., Smith, K.L., Fellows, P.F., Rybachuck, G., Kousoulas, K.G. and Hugh-Jones, M.E. (2003) Bacillus anthracis virulence in Guinea pigs vaccinated with anthrax vaccine adsorbed is linked to plasmid quantities and clonality. Journal of Clinical Microbiology 41, 1212–1218. [74] Andrup, L., Jorgensen, O., Wilcks, A., Smidt, L. and Jensen, G.B. (1996) Mobilization of ‘‘nonmobilizable’’ plasmids by the aggregation-mediated conjugation system of Bacillus thuringiensis. Plasmid 36, 75–85. [75] Kaspar, R.L. and Robertson, D.L. (1987) Purification and physical analysis of Bacillus anthracis plasmids pXO1 and pXO2. Biochemistry Biophysical Research Communications 149, 362– 368. [76] Uchida, I., Sekizaki, T., Hashimoto, K. and Terakado, N. (1985) Association of the encapsulation of Bacillus anthracis with a 60 megadalton plasmid. Journal of General Microbiology 131 (Pt. 2), 363–367. [77] Makino, S.I., Uchida, I., Terakado, N., Sasakawa, C. and Yoshikawa, M. (1989) Molecular characterization and protein analysis of the cap region, which is essential for encapsulation in Bacillus anthracis. Journal of Bacteriology 171, 722–730. [78] Uchida, I., Makino, S., Sasakawa, C., Yoshikawa, M., Sugimoto, C. and Terakado, N. (1993) Identification of a novel gene, dep, associated with depolymerization of the capsular polymer in Bacillus anthracis. Molecular Microbiology 9, 487–496. [79] Makino, S., Watarai, M., Cheun, H.I., Shirahata, T. and Uchida, I. (2002) Effect of the lower molecular capsule released 328 [80] [81] [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 from the cell surface of Bacillus anthracis on the pathogenesis of anthrax. Journal of Infectious Diseases 186, 227–233. Tinsley, E., Naqvi, A., Bourgogne, A., Koehler, T.M. and Khan, S.A. (2004) Isolation of a minireplicon of the virulence plasmid pXO2 of Bacillus anthracis and characterization of the plasmidencoded RepS replication protein. Journal of Bacteriology 186, 2717–2723. Hamon, M.A. and Lazazzera, B.A. (2001) The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Molecular Microbiology 42, 1199–1209. Hamoen, L.W., Kausche, D., Marahiel, M.A., van Sinderen, D., Venema, G. and Serror, P. (2003) The Bacillus subtilis transition state regulator AbrB binds to the 35 promoter region of comK. FEMS Microbiology Letters 218, 299–304. Hamoen, L.W., Venema, G. and Kuipers, O.P. (2003) Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology 149, 9–17. Koehler, T.M. (2002) Bacillus anthracis genetics and virulence gene regulation. Current Topics in Microbiology Immunology 271, 143–164. Saile, E. and Koehler, T.M. (2002) Control of anthrax toxin gene expression by the transition state regulator abrB. Journal of Bacteriology 184, 370–380. Bastos, M.C. and Murphy, E. (1988) Transposon Tn554 encodes three products required for transposition. EMBO Journal 7, 2935–2941. Steichen, C., Chen, P., Kearney, J.F. and Turnbough, C.L. (2003) Identification of the immunodominant protein and other proteins of the Bacillus anthracis exosporium. Journal of Bacteriology 185, 1903–1910. Sylvestre, P., Couture-Tosi, E. and Mock, M. (2003) Polymorphism in the collagen-like region of the Bacillus anthracis BclA protein leads to variation in exosporium filament length. Journal of Bacteriology 185, 1555–1563. Sylvestre, P., Couture-Tosi, E. and Mock, M. (2002) A collagenlike surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Molecular Microbiology 45, 169– 178. Andrup, L., Jensen, G.B., Wilcks, A., Smidt, L., Hoflack, L. and Mahillon, J. (2003) The patchwork nature of rolling-circle plasmids: comparison of six plasmids from two distinct Bacillus thuringiensis serotypes. Plasmid 49, 205–232. Price, L.B., Hugh-Jones, M., Jackson, P.J. and Keim, P. (1999) Genetic diversity of protective antigen genes of Bacillus anthracis. Journal of Bacteriology 181, 2358–2362. Battisti, L., Green, B.D. and Thorne, C.B. (1985) Mating system for plasmid transfer of plasmids among Bacillus anthracis, Baciluus, cereus and Bacillus thuringiensis. Journal of Bacteriology 162, 543–550. Mourez, M., Yan, M., Lacy, D.B., Dillon, L., Bentsen, L., Marpoe, A., Maurin, C., Hotze, E., Wigelsworth, D., Pimental, R.A., Ballard, J.D., Collier, R.J. and Tweten, R.K. (2003) Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proceedings of the National Academy of Sciences of the United States of America 100, 13803–13808. Ackermann, H.W., Roy, R., Martin, M., Murthy, M.R. and Smirnoff, W.A. (1978) Partial characterization of a cubic Bacillus phage. Canadian Journal of Microbiology 24, 986– 993. Stromsten, N.J., Benson, S.D., Burnett, R.M., Bamford, D.H. and Bamford, J.K. (2003) The Bacillus thuringiensis linear double-stranded DNA phage Bam35, which is highly similar to the Bacillus cereus linear plasmid pBClin15, has a prophage state. Journal of Bacteriology 185, 6985–69859. Crickmore, N., Bone, E.J., Williams, J.A. and Ellar, D.J. (1995) Contributions of the indiviual components of the delta-endo- [97] [98] [99] [100] [101] [102] [103] [104] [105] [106] [107] [108] [109] [110] [111] [112] [113] toxin crystal to mosquitocidal activity of Bacillus thuringiensis subsp. israeliensis. FEMS Microbiology Letters 131, 249–2554. Guidi-Rontani, C., Pereira, Y., Ruffie, S., Sirard, J.C., WeberLevy, M. and Mock, M. (1999) Identification and characterization of a germination operon on the virulence plasmid pXO1 of Bacillus anthracis. Molecular Microbiology 33, 407–414. Ason, B., Handayani, R., Williams, C.R., Bertram, J.G., Hingorani, M.M., OÕDonnell, M., Goodman, M.F. and Bloom, L.B. (2003) Mechanism of loading the Escherichia coli DNA polymerase III beta sliding clamp on DNA. Bonafide primer/ templates preferentially trigger the gamma complex to hydrolyze ATP and load the clamp. Journal of Biological Chemistry 278, 10033–10040. Anand, S.P., Mitra, P., Naqvi, A. and Khan, S.A. (2004) Bacillus anthracis and Bacillus cereus PcrA helicases can support DNA unwinding and in vitro rolling-circle replication of plasmid pT181 of Staphylococcus aureus. Journal of Bacteriology 186, 2195–2199. Naqvi, A., Tinsley, E. and Khan, S.A. (2003) Purification and characterization of the PcrA helicase of Bacillus anthracis. Journal of Bacteriology 185, 6633–6639. Wilcks, A., Jayaswal, N., Lereclus, D. and Andrup, L. (1998) Characterization of plasmid pAW63, a second self-transmissible plasmid in Bacillus thuringiensis subsp. kurstaki HD73. Microbiology 144, 1263–1270. Wilcks, A., Smidt, L., Økstad, O.A., Kolstø, A.B., Mahillon, J. and Andrup, L. (1999) Replication mechanism and sequence analysis of the replicon of pAW63, a conjugative plasmid from Bacillus thuringiensis. Journal of Bacteriology 181, 3193–3200. Andrup, L., Smidt, L., Andersen, K. and Boe, L. (1998) Kinetics of conjugative transfer: a study of the plasmid pXO16 from Bacillus thuringiensis subsp. israelensis. Plasmid 40, 30–43. Jensen, G.B., Andrup, L., Wilcks, A., Smidt, L. and Poulsen, O.M. (1996) The aggregation-mediated conjugation system of Bacillus thuringiensis subsp. israelensis: host range and kinetics of transfer. Current Microbiology 33, 228–236. Anisimov, A.P., Lindler, L.E. and Pier, G.B. (2004) Intraspecific diversity of Yersinia pestis. Clinical Microbiology Reviews 17, 434–464. Brown, E.R. and Cherry, W.B. (1955) Specific identification of Bacillus anthracis by means of a variant bacteriophage. Journal of Infectious Diseases 96, 34–39. Stone, R. (2002) Bacteriophage therapy: StalinÕs forgotten cure. Science 298, 728–731. Stone, R. (2002) Bacteriophage therapy: food and agriculture: testing grounds for phage therapy. Science 298, 730. Canchaya, C., Proux, C., Fournous, G., Bruttin, A. and Brussow, H. (2003) Prophage genomics. Microbiology and Molecular Biology Reviews 67, 238–276. Casjens, S. (2003) Prophages and bacterial genomics: what have we learned so far?. Molecular Microbiology 49, 277–300. Desiere, F., McShan, W.M., van Sinderen, D., Ferretti, J.J. and Brussow, H. (2001) Comparative genomics reveals close genetic relationships between phages from dairy bacteria and pathogenic streptococci: evolutionary implications for prophage–host interactions. Virology 288, 325–341. Brussow, H., Canchaya, C. and Hardt, W.D. (2004) Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiology and Molecular Biology Reviews 68, 560. Perna, N.T., Plunkett 3rd, G., Burland, V., Mau, B., Glasner, J.D., Rose, D.J., Mayhew, G.F., Evans, P.S., Gregor, J., Kirkpatrick, H.A., Posfai, G., Hackett, J., Klink, S., Boutin, A., Shao, Y., Miller, L., Grotbeck, E.J., Davis, N.W., Lim, A., Dimalanta, E.T., Potamousis, K.D., Apodaca, J., Anantharaman, T.S., Lin, J., Yen, G., Schwartz, D.C., Welch, R.A. and D.A. Rasko et al. / FEMS Microbiology Reviews 29 (2005) 303–329 [114] [115] [116] [117] [118] [119] [120] [121] [122] [123] [124] Blattner, T. () Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533. Campbell, A.M. (1992) Chromosomal insertion sites for phages and plasmids. Journal of Bacteriology 174, 7495–7499. Schuch, R., Nelson, D. and Fischetti, V.A. (2002) A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418, 884– 889. Ireland, J.A. and Hanna, P.C. (2002) Macrophage-enhanced germination of Bacillus anthracis endospores requires gerS. Infection and Immunity 70, 5870–5872. Volokhov, D., Pomerantsev, A., Kivovich, V., Rasooly, A. and Chizhikov, V. (2004) Identification of Bacillus anthracis by multiprobe microarray hybridization. Diagnostic Microbiology and Infectious Disease 49, 163–171. Zwick, M.E., Macaffee, F., Cutler, D.J., Read, T.D., Ravel, J., Bowman, G.R., Galloway, D.R. and Mateczum, A. (2004) Microarray-based resequencing of multiple Bacillus anthracis isolates. Genome Biology 6, R10. Kroos, L. and Yu, Y.T.N. (2000) Regulation of sigma factor activity during Bacillus subtilis development. Current Opinion in Microbiology 3, 553–560. Warscheid, B. and Fenselau, C. (2004) A targeted proteomics approach to the rapid identification of bacterial cell mixtures by matrix-assisted laser desorption/ionization mass spectrometry. Proteomics 4, 2877–2892. Warscheid, B. and Fenselau, C. (2003) Characterization of Bacillus spore species and their mixtures using postsource decay with a curved-field reflectron. Analytical Chemistry 75, 5618– 5627. Lai, E.M., Phadke, N.D., Kachman, M.T., Giorno, R., Vazquez, S., Vazquez, J.A., Maddock, J.R. and Driks, A. (2003) Proteomic analysis of the spore coats of Bacillus subtilis and Bacillus anthracis. Journal of Bacteriology 185, 1443–1454. Whiting, G.C., Rijpkema, S., Adams, T. and Corbel, M.J. (2004) Characterisation of adsorbed anthrax vaccine by two-dimensional gel electrophoresis. Vaccine 22, 4245–4251. Ariel, N., Zvi, A., Makarova, K.S., Chitlaru, T., Elhanany, E., Velan, B., Cohen, S., Friedlander, A.M. and Shafferman, A. (2003) Genome-based bioinformatic selection of chromosomal Bacillus anthracis putative vaccine candidates coupled with proteomic identification of surface-associated antigens. Infection and Immunity 71, 4563–4579. 329 [125] Oosthuizen, M.C., Steyn, B., Lindsay, D., Brozel, V.S. and von Holy, A. (2001) Novel method for the proteomic investigation of a dairy-associated Bacillus cereus biofilm. FEMS Microbiology Letters 194, 47–51. [126] Oosthuizen, M.C., Steyn, B., Theron, J., Cosette, P., Lindsay, D., von Holy, A. and Brozel, V.S. (2002) Proteomic analysis reveals differential protein expression by Bacillus cereus during biofilm formation. Applied and Environmental Microbiology 68, 2770–2780. [127] Chen, F.C., Shen, L.F., Tsai, M.C. and Chak, K.F. (2003) The IspA proteaseÕs involvement in the regulation of the sporulation process of Bacillus thuringiensis is revealed by proteomic analysis. Biochemical and Biophysical Research Communications 312, 708–715. [128] McNall, R.J. and Adang, M.J. (2003) Identification of novel Bacillus thuringiensis Cry1Ac binding proteins in Manduca sexta midgut through proteomic analysis. Insect Biochemistry and Molecular Biology 33, 999–1010. [129] Avery, O.T., MacLeod, C.M. and McCarty, M. (1944) Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Inductions of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. Journal of Experimental Medicine 79, 137–158. [130] Hayes, W. (1953) Observations on a transmissible agent determining sexual differentiation in Bacti. coli. Journal of General Microbiology 8, 72–88. [131] Lederberg, J., Cavalli, L.L. and Lederberg, E.M. (1952) Sex compatibility in E. coli. Genetics 37, 720–730. [132] Watanabe, T. (1963) Infectious heredity of multiple drug resistance in bacteria. Bacteriological Reviews 27, 87–115. [133] N.R. Smith, R.E. Gordon, F.E. Clark, Aerobic sporeforming bacteria, US Department of Agriculture, Agriculture Monograph No. 16, US Government Printing Office, Washington, DC, 1952. [134] R.E. Gordon, W.C. Haynes, C.H.-N. Pang, The genus Bacillus, Aagriculture Handbook No. 427, United States Department of Argiculture, Agricultural Research Service, Washington, DC, 1973. [135] Rasko, D.A., Myers, G.S. and Ravel, J. (2005) Visualization of comparative genomic analyses by BLAST score ratio BMC. Bioinformatics 6, 2.