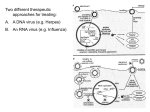
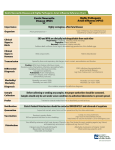
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Western blot wikipedia , lookup
Catalytic triad wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Plant virus wikipedia , lookup
Drug discovery wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Drug design wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
ANRV378-BI78-05 ARI 29 April 2009 ANNUAL REVIEWS Further 10:11 Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. Click here for quick links to Annual Reviews content online, including: • Other articles in this volume • Top cited articles • Top downloaded articles • Our comprehensive search New Antivirals and Drug Resistance Peter M. Colman The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia 3050; email: [email protected] Annu. Rev. Biochem. 2009. 78:95–118 Key Words First published online as a Review in Advance on March 2, 2009 antibody, HIV, influenza The Annual Review of Biochemistry is online at biochem.annualreviews.org This article’s doi: 10.1146/annurev.biochem.78.082207.084029 c 2009 by Annual Reviews. Copyright All rights reserved 0066-4154/09/0707-0095$20.00 Abstract Progress in the discovery of new antiviral medicines is tempered by the rapidity with which drug-resistant variants emerge. A review of the resistance-suppressing properties of four classes of antivirals is presented: influenza virus neuraminidase inhibitors, HIV protease inhibitors, antibodies, and protein-based fusion inhibitors. The analysis supports the hypothesis that the more similar the drug is to the target’s natural ligands, the higher the barrier to resistance. However, other factors, such as entropy compensation and solvent anchoring, might also be exploited for improved drug design. 95 ANRV378-BI78-05 ARI 29 April 2009 10:11 Contents Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. INTRODUCTION AND SCOPE . . . . INFLUENZA VIRUS NEURAMINIDASE . . . . . . . . . . . . . . Neuraminidase Inhibitors . . . . . . . . . . Resistance . . . . . . . . . . . . . . . . . . . . . . . . . The N1 Subtype . . . . . . . . . . . . . . . . . . . THE HIV PROTEASE . . . . . . . . . . . . . . . Inhibitors and Drugs . . . . . . . . . . . . . . . Prodrugs . . . . . . . . . . . . . . . . . . . . . . . . . . Off-Target Resistance . . . . . . . . . . . . . . BIOLOGICALS AS ANTIVIRALS . . . Neutralizing Antibodies . . . . . . . . . . . . Inhibitors of Viral Membrane Fusion. . . . . . . . . . . . . . . . . . . . . . . . . . SUMMARY . . . . . . . . . . . . . . . . . . . . . . . . . . 96 96 97 99 101 102 102 105 106 107 107 109 112 INTRODUCTION AND SCOPE Studies over the past one to two decades have yielded many examples of new drugs and drug candidates for treatment or prevention of viral infections. Most of these discoveries have been aided, if not initiated, by knowledge of the three-dimensional (3D) structure of the target. A large body of structural data accompanies these new medicines and informs an analysis of one of the major issues that bedevils the management of infectious disease, namely the rapid emergence of drug resistance. The differential capacity of drugs to suppress the selection of resistant virus is a property of both the drug and its target. The discovery and clinical experience of neuraminidase inhibitors for influenza and selected drugs for HIV are reviewed in the context of drug resistance. Experience with protein-based antivirals, including emerging structure-based approaches to vaccine design for HIV, are also discussed in this setting. INFLUENZA VIRUS NEURAMINIDASE The neuraminidase inhibitors for treatment and prevention of influenza virus infection are 96 Colman an early example of structure-based drug discovery. Two drugs are approved, a third is in clinical evaluation, and a fourth remains undeveloped. Even though influenza is most commonly an acute infection, clinical drug resistance is observed. The two approved drugs have very different resistance characteristics that can be traced to the detail of their interaction with the conserved catalytic machinery. The neuraminidase of influenza virus (1) is a homotetrameric glycoprotein anchored by a fibrous stalk in the viral membrane. Its primary role in the infectious cycle is to liberate progeny from infected cells, but some roles in mobilizing the virus through mucous secretions has also been described. The enzyme liberates N-acetyl neuraminic acid (Neu5Ac) from α2-3 or α2-6 linkages to galactose, thereby destroying the receptor for the virus. All of the enzymatic and antigenic properties of the protein are associated with the tetrameric globular head, which is liberated when virus is treated with proteolytic enzymes. Ten serologically distinct neuraminidases have been characterized thus far, N1 through N9 associated with type A influenza, and type B neuraminidase from type B influenza viruses. The crystal structures of representatives of the N2 (2, 3), N9 (4), and type B (5) neuraminidases provided the platform for the discovery of the first examples of the neuraminidase inhibitor drug class. Structures of compounds discussed below are depicted in Figure 1. The sequence and structural invariance of the enzyme active site, located on the axis of a β-propeller fold, held the promise for drugs effective against not only all antigenic strains of a particular subtype but also all subtypes and type B (1). The catalytic site has four invaginations accommodating substituents at C2 , C4 , C5 , and C6 on the substrate (6) (Figures 1a and 2a). A cluster of three arginyl residues (R118, R292, R371) encircles the C2 -carboxylate of the substrate, which binds in a twist-boat configuration. A tryptophan residue (W178) engages the C5 -acetyl moiety. A network of hydrogen bond donors and acceptors engage the C6 glycerol and C4 -hydroxyl, as commonly found Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 in carbohydrate-binding proteins and enzymes. Notable among these residues is E276, which forms hydrogen bonds to the C8 and C9 hydroxyls from the glycerol moiety of the substrate. The catalytic nucleophile is believed to be Y406. These amino acids are “preset” for catalysis, because binding of Neu5Ac (or the transition state analog Neu5Ac2en) (Figure 1b) to N2, N9, and subtype B neuraminidases causes no conformational change in the enzyme. Such conditions are likely to be optimal for designing drugs that are truly substrate or transition state mimetics. a HO O N H 4 3 5 2 O 6 HO HO c Neu5Ac2en HO O OH O O N H 1 O- 7 H O HO O- H OH 8 OH HO 9 4-Amino-Neu5Ac2en d Zanamivir H2N O Neuraminidase Inhibitors Replacement of the C4 -hydroxyl moiety of Neu5Ac2en with amino- or guanidino substituents (Figures 1c,d and 2b), designed to optimize chemical and shape complementarity to the enzyme, resulted in approximately 100and 10,000-fold improvements, respectively, in binding potency (7). Zanamivir (4-guanidinoNeu5Ac2en) binds in the active site almost exactly as predicted, but the guanidinium is not hydrogen bonded to E119, rather it forms an ionic contact with it through a stacking interaction (8). Although E119 is conserved in wild influenza viruses, it is evidently not a catalytic residue because different amino acids are found in its place in bacterial and mammalian neuraminidases. Intranasal or inhaled dosing of zanamivir is effective in treatment of influenza in experimental animals (7) and in man (9), but it is poorly distributed when taken orally, even as an ester prodrug. Various attempts to improve the pharmacological properties of zanamivir focused on replacing the glycerol moiety with hydrophobic substituents linked through a carboxamide (10, 11). This study revealed that, although the catalytic machinery of the type A and B neuraminidases appeared almost identical, subtle but significant differences exist in the immediately surrounding structure. These differences manifest themselves as selective binding of Neu5Ac2en analogs, in this case preferentially to type A (12). An example of such a b Neu5Ac +H N 3 O NH2 +HN O O N H O HO O- N H H O HO OH O- H OH HO HO e Diethyl-carboxamide analog of part c O f +H N 3 O N H O Oseltamivir carboxylate O O- +H 3N O N H O- O O N Figure 1 Chemical structures of neuraminidase ligands. glycerol replacement (Figure 1e) is illustrated (Figure 2c). One arm of the diethyl moiety of the carboxamide side chain buries itself in a hydrophobic pocket created by the movement of E276. However, amino acid sequence differences between type A and B neuraminidases conspire to present a higher energy barrier to this modest structural transition in type B than in N2 or N9 neuraminidases. A series of carboxamide analogs of Neu5Ac2en show similar selectivity for type A viruses over type B, making them unattractive clinical candidates (12). www.annualreviews.org • New Antivirals and Drug Resistance 97 ANRV378-BI78-05 ARI 29 April 2009 10:11 a b R224 H274 D151 E276 E119 R292 Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. R118 R371 c d R224 R224 E276 E276 Figure 2 (a) Neu5Ac binds to neuraminidase in a twist-boat configuration. The C2 -carboxylate is surrounded (left to right) by R292, R371, and R118. E119 and D151 are close to the C4 -hydroxyl of the sugar. The C6 -glycerol-binding site includes E276 and R224. H274 is at the far left. The enzyme structure does not change when Neu5Ac binds [Protein Data Bank (PDB) code 1MWE] (124). Molecular figures prepared with PyMOL (125). (b) Zanamivir bound to N9 neuraminidase (PDB code 1NNC). Neu5Ac2en is a transition state analog, and zanamivir is the 4-guanidino derivative of Neu5Ac2en. The protein surface is shaded gray. (c) A carboxamide analog of 4-amino Neu5Ac2en introduces a change in the conformation of the active site at E276, which is now engaged with R224, to create a hydrophobic pocket for the “deep” ethyl group (PDB code 2QWJ). (d ) Oseltamivir carboxylate induces the same conformational change seen in c (PDB code 2QWK). A more radical redesign of the pyran to a carbocyclic scaffold (Figure 1f ) led to the discovery of the orally active neuraminidase inhibitor oseltamivir (13). The active ingredient is generated by the removal of the carboxylic acid 98 Colman ester by host enzymes. The pentyl ether (which now replaces the glycerol) induces similar but subtly different conformational changes in the enzyme to those observed in the Neu5Ac2en carboxamides (Figure 2d ), but now binding ANRV378-BI78-05 ARI 29 April 2009 10:11 to type B neuraminidases is less compromised and leaves open a therapeutic window. A more comprehensive account of the discovery of the neuraminidase inhibitor class is available (14). Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. Resistance The earliest attempts to culture neuraminidase inhibitor-resistant influenza viruses with Neu5Ac2en analogs resulted in viruses with off-target mutations in the receptor-binding site of the hemagglutinin (15), revealing an essential balance between the affinity of the hemagglutinin for the receptor and the efficiency of destruction of the receptor by the neuraminidase. In separate experiments, variants in the catalytic site of the neuraminidase were observed (16). Zanamivir selected viruses with E119G, rationalized by the absence of the ionic interaction with the novel 4-substituent of the drug. E119G has the same enzyme activity as the wild type but is less stable than wild type (17). Other variants at that position, a selected in tissue culture by zanamivir, are E119A and E119D. The glycine and alanine mutants cause loss of binding of zanamivir by up to 1000-fold, and the aspartic acid mutant by up to 2500-fold (18). A resistant virus selected with one of the carboxamide analogs (19) revealed a mutation in one of the three argininyl residues that characterize all known neuraminidases, R292. Substitution to K292 results in some loss of enzyme activity but a large (250- to 1000-fold) loss of binding potency to carboxamide analogs of Neu5Ac2en. Structural studies of carboxamides bound to the R292K variant reveal that E276 does change conformation to allow binding into the hydrophobic pocket (20). The R292K variant most likely acquires resistance by introducing a barrier to the structural transition through which E276 engages R224 and creates the novel hydrophobic binding pocket (Figure 3a). That barrier is a hydrogen bond between E276 and K292, an interaction revealed in the crystal structure of the unliganded R292K variant and not accessible in the wild-type structure. b Carboxamide Oseltamivir carboxylate Wild type Mutant Figure 3 (a) Overlays of the carboxamide of Figure 2c with oseltamivir carboxylate bound to the R292K mutant N9 neuraminidase. Binding to both compounds is compromised by the mutation. A structural rationale is that reduction of carboxamide binding is due to the penalty of inducing the conformational change, whereas for oseltamivir carboxylate, it is due to the failure to create the hydrophobic binding site (PDB codes 2QWG, 2QWH). (b) Overlay of oseltamivir carboxylate bound to wild-type and H274Y mutant N1 neuraminidase. The larger tyrosyl residue in the mutant seems to prevent the movement of E276 that is needed for high-affinity binding (PDB codes 2HU4 and 3C10). www.annualreviews.org • New Antivirals and Drug Resistance 99 ARI 29 April 2009 10:11 The loss of inhibitory potency toward R292K N9 neuraminidase of a panel of neuraminidase inhibitors, including Neu5Ac, Neu5Ac2en, 4-amino-Neu5Ac2en, zanamivir, two Neu5Ac2en carboxamides, and oseltamivir carboxylate, is greater the less the inhibitor resembles the substrate (20). The outlier in similarity to the substrate is oseltamivir carboxylate, for which a 6500-fold loss of binding was determined in the structural background of N9. This analysis supports the principle that drugs that more closely resemble natural substrates or ligands are better equipped to suppress the emergence of a drug-resistant virus. Suppression of such a virus demands a minimal loss of inhibitory activity toward the mutant and a minimal loss of biological activity by the mutant. A drug-resistant virus must be able to maintain its capacity to bind to and (if necessary) process its natural substrates while discriminating between them and the drug. Interestingly, oseltamivir carboxylate does not induce the conformational switch in E276 in R292K mutant N9 neuraminidases (20), unlike the analogous carboxamides (Figure 3a). Oseltamivir carboxylate does not properly engage with the R292K enzyme, the pentyl ether resting “uncomfortably” over the hydrophilic binding site used by the glycerol moiety of the substrate. It may be that the same subtleties that allow oseltamivir carboxylate to retain binding efficacy to wild-type type B neuraminidases are at play. In any event, loss of binding potency of oseltamivir carboxylate to the N2 subtype neuraminidases has been estimated to be as high as 30,000-fold (21). Although experiments in tissue culture are informative, selection pressures in infected patients on drug therapy are very different and can lead to quite different outcomes compared to in vitro studies. Different dosing regimes and routes of administration result in different levels of drug at the site of infection (21a, 21b), and natural clearance mechanisms may be at play in clearing even drug-resistant viruses. A head-tohead comparison of zanamivir and oseltamivir is compromised because zanamivir has been far Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 100 Colman less widely used in the treatment of seasonal influenza than has oseltamivir. Both zanamivir (oral inhalation) and oseltamivir (oral) are prescribed for acute 5day treatment of infected patients. Analysis of matched pairs of virus from 41 patients treated with zanamivir did not reveal any examples of drug-resistant virus (22). Similar studies, with much larger patient numbers, reveal that two percent of oseltamivir-treated individuals shed drug-resistant virus (23). That number is as high as 18% in one pediatric study wherein the dosing is thought to have been suboptimal (24). The R292K mutation is frequently seen in these studies. Other mutations detected in these patients are E119V and N294S. The appearance of drug-resistant viruses in these treated patients does not noticeably compromise the efficacy of treatment, although it may render prophylaxis to immediate contacts ineffective. This may be the way in which amantadine-resistant influenza viruses have taken hold (25, 26). In the N1 antigenic background, the H274Y mutant is selected by oseltamivir and reduces sensitivity to the drug by several hundredfold (27). Unlike a number of other active-site variants, the H274Y virus is transmissible in ferrets (28). Furthermore, this mutant was found in early 2008 in H1N1 human influenza viruses in Europe (29). Of 437 viruses tested, 59 were resistant to oseltamivir by assay or by identification of H274Y, yet there is no evidence that any of the patients from whom samples were obtained had either been treated with oseltamivir or exposed to individuals who had. This observation is causing concern lest this mutation become associated with an avian H5N1 strain and render stockpiled oseltamivir ineffective against an H5N1 pandemic avian influenza. The molecular basis for resistance to H274Y in N1 neuraminidase was described following a structural study of the mutant enzyme bound to both zanamivir and oseltamivir carboxylate (30). Y274 in the mutant prevents E276 from adopting the necessary “switched” conformation demanded by the pentyl ether side chain, whereas substrate and zanamivir are Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 still accommodated (Figure 3b). The 265-fold loss of potency in H274Y (N1) for oseltamivir carboxylate is due to a 10-fold slower association rate and a 25-fold faster dissociation rate. In comparison, the kinetics for binding to zanamivir are essentially the same as for the wild-type H274 (30). Another N1 mutant, N294S, is also resistant to oseltamivir carboxylate by virtue of hydrogen bonding to E276 and restraining the necessary conformational change associated with drug binding (30). Baseline sensitivity of type B viruses to both zanamivir and oseltamivir is lower than for type A viruses, and the emergence in Japan of wild influenza B strains with further reduced sensitivity to neuraminidase inhibitors is of some concern (31, 32). These viruses have been isolated from patients not undergoing treatment with neuraminidase inhibitors, and transmission between individuals seems highly likely. Three such mutants were reported with the following mean IC50 (nM) values against zanamivir and oseltamivir, respectively, D198N (50, 240), I222T (25, 480), and S250G (190, 50). One additional mutant, G402S (50, 280), appeared during treatment with oseltamivir. Wild-type sensitivity (IC50 ) in this study was 6 nM for zanamivir and 72 nM for oseltamivir. Oseltamivir was some 90 times more widely used in Japan than zanamivir in 2004/2005, and it is possible that this has led to the altered drug sensitivity in the communities studied (31). Testing the sensitivity of clade 1 and clade 2 H5N1 neuraminidases to neuraminidase inhibitors has revealed that clade 2 viruses isolated from Indonesia in 2005 are six- to sevenfold less sensitive to oseltamivir than clade 1 viruses (33). It is important to determine if this difference extends to clade 2 viruses isolated elsewehere and to identify its underlying molecular basis, which remains equivocal on the basis of current sequence analysis. No difference in sensitivity to zanamivir was detected in this study. A comparison of resistance properties of oseltamivir and zanamivir must recognize the vast excess of use of the one over the other. A numerical excess of reports of drug resistance to oseltamivir is expected. The most troubling development in resistance is the H274Y mutation in H1N1 viruses from patients apparently naive to any inhibitor. The N1 Subtype Neuraminidase subtypes commonly found in human influenza viruses are N1, N2, and type B. Other subtypes occur in wild and domestic bird populations. H5N1 viruses are the cause of the contemporary highly pathogenic avian influenza, a virus which has prompted many governments around the world to stockpile neuraminidase inhibitors as a first line of defense against its spread in humans. The 3D structure of the N1 subtype is therefore of great interest, even though there were no grounds (on the basis of its amino acid sequence) for believing that it would depart from the structures displayed by N2, N9, and type B neuraminidase. However, the recently described structure of the N1 neuraminidase subtype, together with that of the N4 and N8 subtypes, reveals a new subsite within the enzyme active site (34). As a result of a novel fold for a short loop segment of the structure, amino acids 147–152 in N2 numbering, the pocket adjacent to the C4 -hydroxyl moiety of Neu5Ac is further opened up, inviting exploration by medicinal chemists. Amino acid residues affected by the altered loop conformation include E119 and D151; the former is one of the amino acids targeted by both the guanidinium group of zanamivir and the amino group of oseltamivir carboxylate, and the latter is a putative catalytic residue whose precise function is unknown. The signature amino acid sequence in N1, N4, and N8 that produces this novel architecture has not been identified but seems to include residues beyond the loop itself. When inhibitors were introduced to N1 or N8, either at low inhibitor concentration or with short incubation time, binding was observed in the active site with no change in conformation, i.e., with the enlarged C4 subsite still intact (34). However, higher concentrations of ligand caused the loop to close in on the catalytic site, yielding a structure that www.annualreviews.org • New Antivirals and Drug Resistance 101 ARI 29 April 2009 10:11 appears indistinguishable from those reported earlier for N2, N9, and type B neuraminidase inhibitor complexes. The inhibitory concentrations of zanamivir and oseltamivir carboxylate for N1 are not appreciably weaker than the ∼1 nM values observed for N2 and N9, suggesting that the conformational rearrangement associated with closure of the C4 subsite in N1 comes with little energetic penalty. No function has yet been ascribed to the enlarged C4 subsite of N1. It seems unlikely to be of catalytic significance because of the structural similarity of all complexes of influenza neuraminidases with Neu5Ac2en. Other possible functions for an enlarged catalytic site are altered substrate specificity for the enzyme and even a role in receptor binding, as in the parainfluenza viruses wherein a single active site in the HN protein supports both receptor-binding and catalytic functions (35). No current data support any of these possibilities. In the event that the new site lacks any biological function, targeting it for new antiviral discovery may be of limited therapeutic benefit should resistant viruses rapidly emerge. Nevertheless, exploiting this feature in the design of new neuraminidase inhibitors would further test the hypothesis that greater similarity between a drug and a natural ligand leads to better suppression of resistant virus. Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 THE HIV PROTEASE Although enzymes are generally viewed as favorable drug targets, the protease of HIV poses special problems owing to the dynamic properties of the proteins that control access of substrates to the active site. Nevertheless, protease inhibitors have become an integral component of antiretroviral therapy (36, 37). The cleavage of the Gag and Pol polyproteins of HIV by the viral protease is an essential step in maturation of the virus. The protease is a symmetric dimer with a pair of active-site aspartyl residues (D25, D25 ) in close proximity to the symmetry axes and the scissile bond of the substrate (38–41). The 10 natural polyprotein cleavage sites are remarkably 102 Colman diverse, including at the P1 (four occurrences of F, three of L, and one of Y, M, and N) and P1 (three P, two F, W, Y, L, M, and A) sites (42). Even a palindromic sequence of amino acids is stereochemically asymmetric, and association of the protease dimer with its various substrates is inherently asymmetric (43). Furthermore, access of substrate to the active site requires a movement of the so-called flaps, which are open and loosely structured in the apoenzyme (39, 44) and close over bound substrates and inhibitors (Figures 4 and 5a,b). These features contrast starkly with the influenza neuraminidase wherein the specificity for terminal Neu5Ac is paramount and little or no structural rearrangement of the enzyme occurs during binding. Thus, in addition to the catalytic machinery, functional domains of the protease include those involved in its essential dynamic properties and in its dimerization, and these represent legitimate, if difficult, drug targets. A further distinguishing feature of the protease from the neuraminidase is that its substrates are also the products of a viral gene and hence susceptible to selection pressure. Indeed, protease drug-resistant variants with mutations in both the enzyme active site and in the substrate have been described. Inhibitors and Drugs All but one of the currently registered HIV protease inhibitors are peptidomimetics (37). A vast literature has accumulated on drug resistance-associated mutations, revealing that substitutions in the substrate-binding cleft are often associated with substitutions that determine movement of the flaps, the former compromising substrate (and drug) binding and the latter providing compensating enhanced catalytic efficiency. Thus, the double mutant V82T/I84V directly impacts substrate (Figure 5a) and inhibitor (45) (Figure 5b) interactions in the P1 and P1 sites, and the double mutant M46I/L63P is rate enhancing for many of the natural cleavage events (46). These four mutations alone confer broad resistance to the first generation of protease inhibitors. ANRV378-BI78-05 a ARI 29 April 2009 b Amprenavir H N O O N Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. O H O O OH S O H N N S O H O O H O O O Brecanavir NH2 OH N S O O d CF3 H N O O Darunavir H Tipranavir NH2 OH O c 10:11 H O H N O OH N O S O H O O N O S e f TMC-126 O H O H O H O H N R=H, GS-8373; R=Et, GS-8374 O OH N O S O O H O H O H O H N O OH N S O O O OR P OR O Figure 4 Chemical structures of protease inhibitors. Computational studies support the notion that the compensatory mutation M46I alters the dynamic properties of the flaps (46a). The structural basis for other compensatory mutants (e.g., L90M, which is spatially adjacent to the catalytic aspartate D25) is more subtle. In that case, a computational simulation (46b) fails to capture the experimentally determined poise of nelfinavir bound to the mutant enzyme (46c). Currently, mutations at some 15 residues are characterized in the Stanford HIV database (47, 48) as major resistance mutations for one or more of the approved protease inhibitor drugs. Attention has recently become focused on strategies for improving the performance of the drug class both at inhibiting known drug-resistant strains and at suppressing the emergence of future drug-resistant strains. A structural study of six of the natural substrates, complexed with an inactive (D25N) mutant protease, has led to the idea that specificity is determined primarily by substrate shape (42) and that the envelope defined by this common shape should guide the design of future inhibitors (49, 50). Analysis of the loss of inhibitory potency of five first-generation protease inhibitors to 13 different protease mutants shows some correlation with the volume of the inhibitor lying outside the substrate envelope (50). This study, which formally takes no account of the chemical similarity between the inhibitor and the substrate, is for five peptidomimetic drugs, each with a noncleavable hydroxyethylene linkage in place of the scissile peptide bond (Figure 5b). www.annualreviews.org • New Antivirals and Drug Resistance 103 ANRV378-BI78-05 ARI 29 April 2009 10:11 a P1' pocket residues Catalytic HIV protease dimer aspartates Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ARVLAEAM b P1 pocket residues c Protease surface Protease GS-8373 Core of peptide (VLAE) Amprenavir Figure 5 (a) The HIV protease dimer showing the two catalytic aspartates (mutated to N25 in this structure), the P1, and P1 pocket residues 82 and 84, and the peptide substrate, ARVLAEAM (PDB code 1F7A). (b) Overlay of amprenavir bound to the protease (PDB code 1HPV) with the core of the peptide (VLAE) from panel a. (c) GS-8373, a darunavir analog, modifed with a phosphonate at P1, bound to the protease (trace in green), illustrating the exposure of the phosphonate at the protease surface (shaded green) (PDB code 2I4D). Thus, chemical similarity (as well as shape similarity) to substrate is apparent in all these drugs, and a strong chemical similarity to substrate has previously been qualitatively scored to amprenavir (Figure 4a) (51), the drug favored by the substrate envelope analysis. Tipranavir (Figure 4b), the only nonpeptidomimetic compound (52) currently licensed, also selects resistant variants, albeit slowly and requiring multiple sequence changes (53). 104 Colman Unlike other inhibitors, binding of tipranavir to the protease is almost entirely entropy driven (H -0.7 Kcal/mol; -TS -13.9 Kcal/mol) (54). The mechanism by which it maintains binding affinity to four different drug-resistant mutants, including one selected by tipranavir itself, has been analyzed crystallographically and thermodynamically, showing that the entropic losses that accompany binding to certain mutants are usually either offset by enthalpic Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 gains or, at worst, associated with minimal enthalpic losses. In contrast, in cases where measurements are available for peptidomimetic inhibitors, loss of binding to drug-resistant mutants is largely accounted for by enthalpic losses. Although the structural correlates of this anomalous behavior of tipranavir remain poorly understood, tipranavir binding to the enzyme is characterized by seven direct (as opposed to water-mediated) hydrogen bonds to “conserved” active-site elements (54). The rather different resistance profile of tipranavir to other inhibitors advocates its use in combination therapies. There is not yet any evidence to suggest that tipranavir as a single agent is any more or less effective in suppressing resistance than are the peptidomimetics. The second generation of peptidomimetic protease inhibitors is exemplified by darunavir (Figure 4c), an analog of amprenavir modified in the P2 site (55, 56). Structural studies of darunavir and amprenavir bound to wild-type protease and to a triple mutant (L63P/V82T/I84V) reveal additional interactions between darunavir and the enzyme; these interactions explain its 100-fold tighter binding than amprenavir to wild-type protease. Against the triple mutant, darunavir retains a 33-fold advantage in potency over amprenavir. One feature of the interactions between darunavir and the protease is the extent to which backbone atoms of the protease are engaged, including by the novel bis-tetrahydrofuranylurethane P2 moiety (57). Because of the widely conserved conformation of the protein backbone across diverse protease mutants, a strategy of targeting the backbone promises to suppress drug resistance. Backbone hydrogen bonds to substrate are a feature of the six substrate complexes described above (42), and in this sense, targeting the backbone demands substrate similarity. However, darunavir does make an H-bond to the backbone amide of D30, an interaction not shared with substrates. Darunavir is marginally more effective in inhibiting flap mutants (at positions 48, 50, and 54) than is saquinavir (58). It will be especially interesting to observe the performance of darunavir in the clinic in suppressing drug resistance. Brecanavir (Figure 4d ) is also a derivative of amprenavir/darunavir. It has a P1 site extension terminating with a thiazole, which structurally closely overlays with the phosphonate of GS8374 (see below) (59). Brecanavir also retains potency against a broad spectrum of protease inhibitor-resistant viruses. In vitro brecanavir selects mutants in the protease active site, although high-level resistance was only observed in the context of A28S, a mutation that reduces viral replication efficacy (60). Despite this encouraging profile with respect to potency and resistance, the development of brecanavir was halted in December 2006 because of formulation problems, another reminder of the many competing properties that are sought in drug candidates. Prodrugs One way to improve the therapeutic profile of a drug is to alter its distribution in favor of the site of action of the drug target, in this case inside an infected cell. Increasing the effective concentration of the drug might suppress mutants that would otherwise be viable. Thus, attachment to a protease inhibitor scaffold of a charged phosphonate, masked by a chemical moiety that is selectively removed by intracellular enzymes, should improve the retention of the compound inside cells (61). Pursuit of this strategy, based on derivitizing the P1 position of TMC-126 (Figure 4e) (an analog of amprenavir and darunavir), led to the unexpected finding that a phosphonate-containing inhibitor, GS-8373 (Figure 4f ), has an improved binding profile to a broad range of inhibitor-resistant protease variants. Crystallographic analysis reveals that the phosphonate moiety of GS-8373 projects into the solvent (Figure 5c), on a track adjacent to but not overlapping with the path used by substrates, where it is loosely ordered with few if any specific interactions with the protease. The diethyl ester prodrug (GS-8374) (Figure 4f ) is www.annualreviews.org • New Antivirals and Drug Resistance 105 ARI 29 April 2009 10:11 an equally potent inhibitor in vitro, supporting the conclusion that the phosphonate makes no specific interactions with the protease. The performance of the two compounds in antiviral assays is as predicted: The charged phosphonate is inactive, and the neutral form shows equal or better activity to TMC-126. Isothermal titration calorimetry of GS-8373, GS-8374, TMC-126, and amprenavir to two mutant proteases (M46I/I47V/I50V and I84V/L90M) showed that the phosphonates retain activity to the mutants, whereas the nonphosphonates suffer a 10- to 40-fold loss of activity. A similar trend was observed for phosphonatederivatized atazanavir. In all cases studied, the calorimetric data show enthalpic losses in the binding interaction to mutants compared with wild type, but entropic compensation for these losses is always greater in the phosphonatecontaining inhibitors thereby minimizing the loss of binding potency to the mutants. A detailed structure analysis shows that the poise of the TMC-126 core is identical in wild type and in the I84V/L90M mutant. In contrast, a small but significant difference is observed for GS8374 in wild type compared to mutant, suggesting it is more adaptable to its binding site than is TMC-126 and could be considered anchored by the solvent (61). This approach is suggested as a way forward for suppression of drug-resistant virus (61, 62). Attempts to culture virus in the presence of various phosphonate-containing protease inhibitors were unsuccessful, but parallel studies with TMC-126 generated an L10F/A28S/M46I/I50V mutant. These results seem at odds with the substrate similarity principle for suppression of drug resistance. The phosphonate moiety fails the chemicalsimilarity-to-substrate test and occupies a volume beyond the substrate envelope, as defined above (50). Of course, the natural substrate is the polyprotein, which does extend beyond the conventionally defined substratebinding cleft. Perhaps the solvent-anchored moiety is mimicking these distal elements of the substrate and/or targeting regions of the protease structure that are essential for the Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 106 Colman dynamic incorporation of substrate into the active site, in which case resistant mutants may be unviable. Its near neighbors on the protein are G52, F53, and P81, and mutation at G52 or P81 has not yet been reported (47). If these residues are somehow functionally important, then the approach for HIV holds great promise. Exemplifying the generalization of this approach to other targets will be watched with great interest. Off-Target Resistance Off-target resistance to protease inhibitors has also been reported. Mutations in cleavage sites of the Gag polyprotein have previously been viewed as “rescue mutants” in the context of protease mutations that directly affect drug binding (63). However, some studies have detected resistance to protease inhibitors without concomitant mutations in the protease that affect drug binding (64), suggesting off-target mutations. More recently in vitro selection with the protease inhibitor RO033-4649 has resulted in drug-resistant variants possessing Gag mutations with no protease mutations (65); the Gag mutants were associated with increased processing efficiency for one of the intermediate products (NCp6). It was also shown that the NC/p1 cleavage site mutant A431V could confer fourfold resistance to ritonavir. Evidently, increasing the susceptibility of the polyprotein to protease processing is a novel and bona fide mechanism by which HIV acquires resistance to protease inhibitors (65). Longer-term culturing of virus with GS-8374 led to a 15-fold loss of drug sensitivity owing to mutations in the Gag polyprotein. The only protease mutant detected was K41R, a mutation outside the peptide-binding tunnel but close to the peptide’s entry and exit points. However, this mutation alone does not alter drug sensitivity. The observed 15-fold loss of efficacy could be mapped entirely to the Gag mutations (66). One unmistakable trend in current protease inhibitor drug discovery is toward compounds with improved resistance properties. Molecules with increased substrate similarity, both in shape and chemistry, have improved Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 performance in inhibiting extant drug-resistant strains and in suppressing the emergence of new ones (50, 51). Nevertheless, inhibitors, such as GS-8374 (61) and tipranavir (54), that display obvious departures from substrateness also score well in resistance properties. It remains to be seen whether GS-8374 is additionally targeting some other essential functionality of the protease, such as its dynamic properties. The appearance of off-target resistance may pose an ultimate barrier to the degree to which a protease inhibitor can truly suppress the selection of drug-resistant virus. BIOLOGICALS AS ANTIVIRALS There are few examples of registered proteinbased antivirals, but more are expected in future. Here the resistance-suppressing prospects of such antiviral agents are considered. Neutralizing Antibodies Neutralizing antibodies, raised through vaccination or prior infection, are highly successful antiviral agents. Of course, viral strain variation can give rise to resistance to antibody. Following the early structural studies of the structure and antigenicity of influenza virus antigens (2, 67) and human rhinoviruses (68), a large body of data has accumulated around the question of antigenic escape from neutralizing antibody. Landmark studies, using monoclonal antibodies to select “monoclonal” variants (69), showed that single amino acid sequence changes sufficed to abolish binding of the antigen to the antibody, and subsequent structural studies showed that the structural consequences of these single amino acid sequence changes were indeed only local (70, 71). Later, it was realized that certain amino acid sequence substitutions could be tolerated within the antibody-antigen interface (72), that two different antibodies could engage a common epitope in chemically unrelated ways (73), and that mutations in an epitope could have quite different effects on the binding of two antibodies sharing that epitope (74). Functionally, essential sites of viruses appear susceptible to antibody surveillance, but a number of mechanisms operate to avoid detection. In the case of the influenza antigens, the enzyme site of the neuraminidase presents a surface that is smaller than a typical footprint of an antibody-binding site (2, 75). Carbohydrate masking and conformational gymnastics feature as protection mechanisms of HIV gp160 (76). Even when a functional site is exposed, a thought experiment suggests that only a special subset of antibodies, i.e., those that see the site in the same way as does the natural ligand, would be truly broadly neutralizing (77). Vaccination against influenza virus requires annual matching of vaccine strains to circulating viral strains. Despite an improved understanding of global circulation and evolution of type A influenza viruses (78), mismatches still occur (79). For other viral diseases (smallpox, measles, mumps, hepatitis B), vaccination is extraordinarily successful. For HIV, the large number of recorded strains suggests that vaccination to raise neutralizing antibody will fail to protect against nonhomologous strains. However, the characterization of a number of broadly neutralizing antibodies for HIV (80) has raised the prospect of attempting to elicit just such a response via vaccination. Four such antibodies have now been well characterized: b12 binds to the CD4-binding site on gp120, 2G12 binds a cluster of oligomannose residues, and 2F5 and 4E10 bind to membrane proximal sites on gp41. The International AIDS Vaccine Initiative has established a Neutralizing Antibody Consortium, which is pursuing the crystal structures of antibody complexes with components of gp160 as a basis for rational vaccine design (81). Already, crystal structures for these four broadly neutralizing antibodies have been determined. Antibody b12 is one of the few full-length immunoglobulin structures to have yielded to crystallographic analysis. It has an elongated CDR H3 loop, which is thought to insert into the recessed CD4-binding site on gp120 (82). A structure of the Fab in complex with a peptide designed to mimic the discontinuous epitope www.annualreviews.org • New Antivirals and Drug Resistance 107 ARI 29 April 2009 10:11 of b12 disproved the design hypothesis, indicating how difficult is the design of immunogens (83). Antibody 2G12 has a unique domain swap generating a dimeric structure with two normal antigen-binding sites, (each comprising one VL and one VH domain) flanking one unusual binding site at the interface of the two VH domains (84). Cocrystallization with mannosecontaining carbohydrates confirms that the normal sites each bind one oligosaccharide and that the unusual site binds two. Oligomannose occurs on gp160 of most HIV strains. Structural studies of antibody 2F5 complexed with cognate peptides from the membrane proximal region of gp41 (85, 86) reveal that the apex of the CDR H3 loop of the antibody is formed by four hydrophobic residues, L, F, V, and I. These amino acids play little part in binding the peptide, and they are exposed in a way that may promote association with a membranetethered peptide. Nonconservative mutation of the phenylalanine residue in the apex decreases binding to both the peptide and to gp41 by up to 10-fold, but severely (100-fold) compromises the neutralizing effect of 2F5 against otherwise sensitive HIV strains (87). Antibody 4E10 also has a long CDR H3 loop, which extends beyond the contacting interface with a 13-residue helical peptide (88). Here, the apical CDR H3 residue is W, again so oriented that it could contact the viral membrane when 4E10 binds to virus. Crystal structures of 4E10 Fab, bound to three related designed peptides, have led to a better definition of the epitope and of ways of stabilizing its structure for incorporation into a candidate immunogen (89). Collectively, these structural data provide a good launching pad for designing structures that might elicit broadly neutralizing antibodies. However, the critically important structure of the natural immunogen, gp160, still remains unknown. The Neutralizing Antibody Consortium’s strategy is to extend the panel of known broadly neutralizing antibodies and characterize their epitopes, determine the 3D structure of functional HIV glycoproteins with and without these antibodies bound, and use this information to rationally design immunogens (90). Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 108 Colman The recent NIAID HIV Vaccine Summit in March 2008 listed as two of its nine highest research priorities the determination of the 3D structure of the envelope trimer and finding a way to elicit broadly neutralizing antibody (91). In combating a virus where strain variation arises readily through error-prone replication, an antibody that is capable of binding to all functional strains of the virus would need to have the very special properties of binding exclusively to a functional site and engaging that site in a chemically similar way to the functional binding partner of the virus (77). This goal might be achieved with an appropriate polyclonal response to an epitope representing, say, the receptor-binding site on a virus or the viral fusion machinery. Currently only one monoclonal antibody is registered as an antiviral agent. Palivizumab (Synagis®) is a humanized mouse monoclonal antibody approved for antiviral prophylaxis in pediatric populations susceptible to respiratory syncitial virus (RSV). Antibody-resistant viruses have been selected in tissue culture and then tested for prophylactic efficacy in cotton rats (92). Three variants in the RSV Fusion (F) protein were selected in vitro, K272M, K272Q, and N268I. The structure of the RSV F protein is known only by homology with the paramyxovirus F proteins (93). Interestingly, in both the pre- and postfusion structures of the F protein (94–96), these amino acids are located in the surface between two helical segments. Infection of animals with N268I virus was still blocked by palivizumab, but with K272Q, the virus was not. The F protein mutant K272Q was also selected in vivo (97), and in tissue culture, the K272M virus grows less well than the N268I virus (98). An affinity-matured form of palivizumab is in development. It is not known whether clinical failures of palivizumab prohylaxis in premature infants (99) were the result of infection by antibody-resistant RSV strains. There is considerable interest in the use of monoclonal antibody therapy for hepatitis C virus, especially in liver transplant patients (99a). These developments stand to benefit ANRV378-BI78-05 ARI 29 April 2009 10:11 from experiences in other settings of viral infection. Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. Inhibitors of Viral Membrane Fusion Current understanding of the molecular basis for the fusion of a viral membrane with a target cell membrane stems from the description of the structure of the influenza virus hemagglutinin in its pre- and postfusion conformations (100). In one other system, the paramyxoviruses, comparable information is available on the structures of the pre- and postfusion forms of the fusion protein (96, 101). For HIV, a structure for gp160 remains elusive, but numerous studies of the postfusion conformation of gp41 have informed the exploitation of the fusion machinery as a drug target. Early studies on the antiviral properties of peptides, spanning putative helical regions of HIV gp41(102–104), have their basis in the observation that membrane fusion follows the formation of a structure often referred to as a six-helix bundle (105, 106). (Figure 6a). This helical bundle is believed to be formed via a pathway in which one intermediate is a trimeric coiled-coil structure comprising helical region 1 (or HR1), which can associate with the target membrane via a “fusion peptide” at its N-terminal end. A second helical region (HR2) from the C terminus of the fusion protein engages with the trimeric coiled coil in an antiparallel complex such that its C terminus, inserted in the viral membrane, is at the same end of the helical bundle as the target membrane. Thus, the putative intermediate structure, the trimeric coiled coil of HR1 peptides, has been viewed as a potential drug target for disruption of the fusion-competent conformer. Enfuvirtide (T-20) is the first drug to validate this approach. It is an oligopeptide, comprising residues 127–162 of the HIV fusion protein gp41 (Figure 6b). This sequence overlaps with HR2 (residues 117–154) as defined in structural studies (106), and its mode of action is generally believed to be prevention of the formation of the helical bundle by competition with an identical sequence on the virus. Such a drug seems to meet the requirement of closely resembling the natural substrate, and yet the barrier to emergence of enfuvirtide-resistant virus in the clinic is low (107). The earliest studies of resistance to enfuvirtide in tissue culture (108) revealed drug-resistant mutations in HR1, in the peptide sequence G36-I37-V38, but no compensating mutations were observed in HR2, despite the fact that G36 and V38 face residues K144 and N145 in the outer HR2 helix (106). Other studies suggest that compensating mutations might occur elsewhere in the envelope protein (109). Resistant variants selected in the clinic map to this same region (36–45) of HR1 (107). Common resistance mutations are G36 to D, S, E, and V and V38 to A, G, E, and M. Loss of binding of enfuvirtide to corresponding mutants displayed in the HR1 region of gp41 correlates with loss of antiviral activity (110), supporting the proposed mode of action (Figure 6c). However, other studies show that enfuvirtide only weakly inhibits formation of the sixhelix bundle, at least when compared with peptides representing different segments of HR2 (111), suggesting that the mode of action of enfuvirtide may be more complex than previously thought. The structure of the HR1 trimeric coiled coil presents a prominent pocket-shaped feature into which amino acids W117, W120, D121, and I124 from HR2 are inserted when the helical bundle is formed (Figure 6c). Such a cavity is an enticing target for drug discovery, and several attempts are underway to exploit it, including the use of D-peptides (112, 113). A discussion of organic antagonists of fusion is beyond the scope of this article. The cavity on the HR1 trimer is toward its C-terminal end (near G61), and the residues of HR2 that insert into it are not present in enfuvirtide (Figure 6b). Considering the HR1 coiled coil as the drug target, at its N-terminal end are the mutations conferring resistance to enfuvirtide, and at its C-teminal end is the cavity (Figure 6c). A study of the antiviral properties of a number of overlapping HR2 peptides www.annualreviews.org • New Antivirals and Drug Resistance 109 ANRV378-BI78-05 ARI 29 April 2009 10:11 a HR2 HR1 HR1 HR2 HR1 HR2 Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. b 77 HR1 30 KLYREVALIRAQLQKIGWVTLQLLHQQAEIARLLNNQQQVIGSLLQRA — fusion-peptide 117 154 WMEWDREINNYTSLIHSLIEESQNQQEKNEQELLELDK WASLWNWFNISNWLWYIR HR2 Links HR2 to membrane anchor Enfuvirtide Literature peptides T-20 T-1249 T-2635 SFT YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASLWEWF TTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAALREL WIEWEREISNYTNQIYEILTESQNQQDRNEKDLLE c G36 mutation site V38 mutation site Figure 6 (a) The postfusion state of the assembly of helical regions one and two (HR1 and HR2) in gp41 (PDB code 1ENV). The trimeric nature of the assembly is illustrated by its coloring ( green, cyan, and magenta), and the ribbon drawing of the magenta subunit is overlayed with the atomic structure in stick format. The N-terminal end of HR1 and the C-terminal end of HR2 are to the right as in panel b. (b) The first and second lines are the amino acid sequences of HR1 and HR2, with their approximate structural alignment from PDB code 1ENV. (c) The six-helix bundle of gp41 with surfaces colored as in (a) and with HR2 of the magenta subunit shown as a coil and highlighting ( yellow surface) the enfuvirtide-resistant mutation sites G36 and V38. Also shown in stick format is the pocket-binding motif at the N-terminal end of HR2 (PDB code 1ENV). including 127–162 (enfuvirtide), 122–157, and 117–152 has recently shown that the middle sequence is some 100-fold less potent than either of the other two (111). This suggests that the 110 Colman HR2 helix has distinct domains of interaction with HR1, including with the cavity at one end and with lipids or the fusion peptide at the other end. Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 T-1249 is a second generation peptide, again derived from amino acid sequences comprising and adjacent to HR2 (114). The sequence is a chimera derived from HIV-1, HIV-2, and SIV and displays a pocket-binding motif (see above) at its N-terminal end, although the location of this motif is displaced by one heptad from its position in the gp41 sequence (Figure 6b). Drugresistant virus has been selected both in vitro and in vivo, displaying HR1 mutants (residues 37 and 38) that are cross-resistant to T-20 (115). A subsequent study in patients already treated with enfuvirtide generated mutants in both HR1 and HR2 (116). Clinical development of T-1249 has been discontinued (117). A third-generation peptide, T-2635 (Figure 6b), has also been reported with a span similar to the HR2 helix but containing engineered sequences to enhance helicity and stability (118). Curiously, V38 mutants selected with enfuvirtide or T-1249 remain susceptible (some even more susceptible than wild type) to T-2635 (119), even though the sequences on the peptides adjacent to V38 are essentially identical. The structural basis for these observations is unknown. Perhaps the modes of interaction of enfuvirtide, T-1249, and T-2635 with HR1 are not identical. Perhaps the additional interaction energy deriving from the pocket-binding motif at the N-terminal end of T-2635 allows efficacious binding even in the face of V38 mutations. It will be interesting to see whether the nonnative sequences in T-2635 give rise to a novel panel of resistant viruses. Sifuvirtide, another third-generation peptide (Figure 6b), is similar in span and sequence to T-2635 and contains engineered elements for stability, potency, and pharmacokinetics (120). It lacks the C-terminal decapeptide of enfuvirtide, which may explain its failure to interact with lipid membranes. Like T-2635, it prevents replication of enfuvirtide-resistant HIV strains, and it was safe and well-tolerated in a Phase 1a study (120). Its profile in suppressing the emergence of drug-resistant virus is yet unknown. A complementary drug target is HR2 itself, but until recently, attempts to exploit this target with peptides related to HR1 have been frustrated by poor solubility of the relevant sequences. In isolation, HR1 is expected to exist as a trimeric coiled coil. Commencing with an HR1 sequence including residues 29 through 79 (T-865), an engineered peptide, which includes the core “a” and “d” residues of the heptad from HR1 of RSV, has improved stability and antiviral activity (118). Passaging virus in the presence of a related HR1 trimeric sequence selected variants with mutations in HR2, supporting the model for the mode of action of the HR1 peptides. Once again, it will be interesting to see whether the use of nonnative sequences in HR1 lowers the barrier for drug resistance to emerge. A novel way to target HR2 and to overcome some of the solubility issues with HR1 trimers is with the peptide known as 5-helix (121). This construct contains the sequence HR1-HR2-HR1-HR2-HR1 connected with linkers and assembles into a 5-helix bundle (122), presenting a binding site for one HR2 helix (Figure 6c). 5-helix is inhibitory of a number of HIV variants at nanomolar levels (121). Kinetic studies suggest that, during viral infection, the HR2 helix is accessible for only a few seconds, underscoring the importance of the association kinetics of any agent that seeks to capture it (123). The resistance profile of 5-helix is unknown. The early structural insights into viral membrane fusion are the foundation for the fusioninhibitor class of HIV drugs. The pattern of drug-resistant mutations generally fits a model with the mode of action through disruption of formation of the fusion-competent conformer of gp41. Nevertheless, a molecular interpretation of the enfuvirtide-selected resistance mutations still lacks a sound structural basis. No examples of structures of drug bound to its target, nor to a drug-resistant target, are currently available. That aside, we remain confronted with the fact that the native sequence from HR2 and the adjacent membrane proximal region represented in the drug select for resistance. Features of enfuvirtide that are departures from the “natural ligand” include that it is monomeric, is not covalently associated www.annualreviews.org • New Antivirals and Drug Resistance 111 ANRV378-BI78-05 ARI 29 April 2009 10:11 with HR1, and is not representative of the entire binding surface to the HR1 coiled coil. Future analysis of the second- and third-generation HR2-based peptides and of the emerging opportunities with HR1-based peptides will further inform approaches to raising the resistance barrier to this important drug class. SUMMARY Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. Target sites for antivirals should be functional sites. The failure of antibody-mediated immunity to combat viral strain variation is usually a failure of the antibody to target a functional site. Notwithstanding the enormous impact that protease inhibitors have made as part of the anti-HIV armamentarium, a second-order preference for the target might be that its function should be directed at host molecules and not at virus-encoded molecules, which are themselves subject to selection. Minimizing the chemical differences between the target’s natural ligands and the drug will raise the barrier to the emergence of mutated targets that can distinguish between the two. Multicomponent therapy will continue as a prominent strategy for suppressing the emergence of drug-resistant variants, but increased attention will be paid to optimizing the resistance-suppressing characteristics of the individual components. The principles discussed are clearly not restricted to antiviral drug discovery. SUMMARY POINTS 1. The barrier to emergent resistance is a property of the drug and its target. 2. Very subtle chemical changes in the drug can radically alter that barrier. 3. The drug-binding site is best confined to functionally essential elements. 4. Stereochemical similarity between the drug and natural ligands is desirable. FUTURE ISSUES 1. Further review is needed of extant resistance data for generic trends to inform new drug discovery. 2. Monitoring the performance of new drugs with purportedly higher barriers to resistance should continue. DISCLOSURE STATEMENT The author owns shares in Biota Holdings Ltd., which receives royalties from GlaxoSmithKline on sales of Relenza. ACKNOWLEDGMENTS I thank Brian Smith for helpful discussions and the NHMRC (Australia) for support. LITERATURE CITED 1. Colman PM. 1994. Influenza virus neuraminidase: structure, antibodies, and inhibitors. Protein Sci. 3:1687–96 112 Colman Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 2. Colman PM, Varghese JN, Laver WG. 1983. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 303:41–44 3. Varghese JN, Laver WG, Colman PM. 1983. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature 303:35–40 4. Baker AT, Varghese JN, Laver WG, Air GM, Colman PM. 1987. Three-dimensional structure of neuraminidase of subtype N9 from an avian influenza virus. Proteins 2:111–17 5. Burmeister WP, Ruigrok RW, Cusack S. 1992. The 2.2 Å resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 11:49–56 6. Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA, Colman PM. 1992. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 14:327–32 7. von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, et al. 1993. Rational design of potent sialidasebased inhibitors of influenza virus replication. Nature 363:418–23 8. Varghese JN, Epa VC, Colman PM. 1995. Three-dimensional structure of the complex of 4-guanidinoNeu5Ac2en and influenza virus neuraminidase. Protein Sci. 4:1081–87 9. Hayden FG, Treanor JJ, Betts RF, Lobo M, Esinhart JD, Hussey EK. 1996. Safety and efficacy of the neuraminidase inhibitor GG167 in experimental human influenza. JAMA 275:295–99 10. Sollis S, Howes P, Smith P, Cherry P, Bethell R. 1996. Novel inhibitors of influenza sialidase related to GG167. Synthesis of 4-amino and guanidino-4H-pyran-2-carboxylic acid-6-propylamides; selective inhibitors of influenza A sialidase. Bioorg. Med. Chem. Lett. 6:2931–36 11. Smith PW, Whittington AR, Sollis SL, Howes PD, Taylor NR. 1997. Novel inhibitors of influenza sialidases related to zanamivir. Heterocyclic replacements of the glycerol sidechain. Bioorg. Med. Chem. Lett. 7:2239–42 12. Taylor NR, Cleasby A, Singh O, Skarzynski T, Wonacott AJ, et al. 1998. Dihydropyrancarboxamides related to zanamivir: a new series of inhibitors of influenza virus sialidases. 2. Crystallographic and molecular modeling study of complexes of 4-amino-4H-pyran-6-carboxamides and sialidase from influenza virus types A and B. J. Med. Chem. 41:798–807 13. Kim CU, Lew W, Williams MA, Liu H, Zhang L, et al. 1997. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 119:681– 90 14. Colman P. 2006. Anti-influenza drugs from neuraminidase inhibitors. In Structure-Based Drug Discovery: An Overview, ed. RE Hubbard, pp. 193–218. Cambridge, UK: RSC Publ. 15. McKimm-Breschkin JL, Blick TJ, Sahasrabudhe A, Tiong T, Marshall D, et al. 1996. Generation and characterization of variants of NWS/G70C influenza virus after in vitro passage in 4-amino-Neu5Ac2en and 4-guanidino-Neu5Ac2en. Antimicrob. Agents Chemother. 40:40–46 16. Blick TJ, Tiong T, Sahasrabudhe A, Varghese JN, Colman PM, et al. 1995. Generation and characterization of an influenza virus neuraminidase variant with decreased sensitivity to the neuraminidase-specific inhibitor 4-guanidino-Neu5Ac2en. Virology 214:475–84 17. McKimm-Breschkin JL, McDonald M, Blick TJ, Colman PM. 1996. Mutation in the influenza virus neuraminidase gene resulting in decreased sensitivity to the neuraminidase inhibitor 4-guanidinoNeu5Ac2en leads to instability of the enzyme. Virology 225:240–42 18. McKimm-Breschkin JL. 2000. Resistance of influenza viruses to neuraminidase inhibitors–a review. Antivir. Res. 47:1–17 19. McKimm-Breschkin JL, Sahasrabudhe A, Blick TJ, McDonald M, Colman PM, et al. 1998. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2enderived inhibitors. J. Virol. 72:2456–62 20. Varghese JN, Smith PW, Sollis SL, Blick TJ, Sahasrabudhe A, et al. 1998. Drug design against a shifting target: a structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure 6:735–46 21. Tai CY, Escarpe PA, Sidwell RW, Williams MA, Lew W, et al. 1998. Characterization of human influenza virus variants selected in vitro in the presence of the neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 42:3234–41 www.annualreviews.org • New Antivirals and Drug Resistance 113 ARI 29 April 2009 10:11 21a. Cass LM, Efthymiopoulos C, Bye A. 1999. Pharmacokinetics of zanamivir after intravenous, oral inhaled or intranasal administration to healthy volunteers. Clin. Pharmacokinet. 36(Suppl. 1):1–11 21b. He G, Massarella J, Ward P. 1999. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin. Pharmacokinet. 37:471–84 22. Barnett JM, Cadman A, Gor D, Dempsey M, Walters M, et al. 2000. Zanamivir susceptibility monitoring and characterization of influenza virus clinical isolates obtained during phase II clinical efficacy studies. Antimicrob. Agents Chemother. 44:78–87 23. Jackson HC, Roberts N, Wang ZM, Belshe R. 2000. Management of influenza: use of new antivirals and resistance in perspective. Clin. Drug Investig. 20:447–54 24. Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, et al. 2004. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 364:759–65 25. Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, et al. 2005. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 366:1175–81 26. Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. 2006. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 295:891–94 27. Gubareva LV, Webster RG, Hayden FG. 2001. Comparison of the activities of zanamivir, oseltamivir, and RWJ-270201 against clinical isolates of influenza virus and neuraminidase inhibitor-resistant variants. Antimicrob. Agents Chemother. 45:3403–8 28. Herlocher ML, Truscon R, Elias S, Yen HL, Roberts NA, et al. 2004. Influenza viruses resistant to the antiviral drug oseltamivir: transmission studies in ferrets. J. Infect. Dis. 190:1627–30 29. Lackenby A, Hungnes O, Dudman SG, Meijer A, Paget WJ, et al. 2008. Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Eurosurveillance 13. Online http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8026 30. Collins PJ, Haire LF, Lin YP, Liu J, Russell RJ, et al. 2008. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 453:1258–61 31. Hatakeyama S, Sugaya N, Ito M, Yamazaki M, Ichikawa M, et al. 2007. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA 297:1435–42 32. Moscona A, McKimm-Breschkin J. 2007. News about influenza B drug resistance that cannot be ignored. JAMA 297:1492–93 33. McKimm-Breschkin JL, Selleck PW, Usman TB, Johnson MA. 2007. Reduced sensitivity of influenza A (H5N1) to oseltamivir. Emerg. Infect. Dis. 13:1354–57 34. Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, et al. 2006. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443:45–49 35. Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, et al. 2004. Structure of the haemagglutininneuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335:1343–57 36. Wlodawer A. 2002. Rational approach to AIDS drug design through structural biology. Annu. Rev. Med. 53:595–614 37. De Clercq E. 2007. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 6:1001–18 38. Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, et al. 1989. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 337:615–20 39. Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, et al. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616–21 40. Lapatto R, Blundell T, Hemmings A, Overington J, Wilderspin A, et al. 1989. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature 342:299– 302 41. Wlodawer A, Erickson JW. 1993. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 62:543–85 42. Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. 2002. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 10:369–81 43. Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. 2000. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 301:1207–20 Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 114 Colman Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 44. Heaslet H, Rosenfeld R, Giffin M, Lin YC, Tam K, et al. 2007. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. D Biol. Crystallogr. 63:866–75 45. Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, et al. 1995. Crystal structure of HIV-1 protease in complex with Vx-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 117:1181–82 46. Schock HB, Garsky VM, Kuo LC. 1996. Mutational anatomy of an HIV-1 protease variant conferring cross-resistance to protease inhibitors in clinical trials. Compensatory modulations of binding and activity. J. Biol. Chem. 271:31957–63 46a. Piana S, Carloni P, Rothlisberger U. 2002. Drug resistance in HIV-1 protease: flexibility-assisted mechanism of compensatory mutations. Protein Sci. 11:2393–2402 46b. Ode H, Neya S, Hata M, Sugiura W, Hoshino T. 2006. Computational simulations of HIV-1 proteases multidrug resistance due to nonactive site mutation L90M. J. Am. Chem. Soc. 128:7887–94 46c. Kozisek M, Bray J, Rezacova P, Saskova K, Brynda J, et al. 2007. Molecular analysis of the HIV-1 resistance development: enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J. Mol. Biol. 374:1005–16 47. Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, et al. 2007. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 21:215–23 48. Shafer RW, Schapiro JM. 2008. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 10:67–84 49. Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. 2003. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J. Virol. 77:1306–15 50. Chellappan S, Kairys V, Fernandes MX, Schiffer C, Gilson MK. 2007. Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins 68:561–67 51. Colman PM, Smith BJ. 2003. Specificity and promiscuity in protein-ligand and protein-protein interactions. Aust. J. Chem. 56:763–67 52. Thaisrivongs S, Skulnick HI, Turner SR, Strohbach JW, Tommasi RA, et al. 1996. Structure-based design of HIV protease inhibitors: sulfonamide-containing 5,6-dihydro-4-hydroxy-2-pyrones as nonpeptidic inhibitors. J. Med. Chem. 39:4349–53 53. Doyon L, Tremblay S, Bourgon L, Wardrop E, Cordingley MG. 2005. Selection and characterization of HIV-1 showing reduced susceptibility to the nonpeptidic protease inhibitor tipranavir. Antivir. Res. 68:27–35 54. Muzammil S, Armstrong AA, Kang LW, Jakalian A, Bonneau PR, et al. 2007. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J. Virol. 81:5144–54 55. King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. 2004. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J. Virol. 78:12012–21 56. Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, et al. 2005. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem. 48:1813–22 57. Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. 2008. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res. 41:78–86 58. Liu F, Kovalevsky AY, Tie Y, Ghosh AK, Harrison RW, Weber IT. 2008. Effect of flap mutations on structure of HIV-1 protease and inhibition by saquinavir and darunavir. J. Mol. Biol. 381:102–15 59. Miller JF, Andrews CW, Brieger M, Furfine ES, Hale MR, et al. 2006. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett. 16:1788– 94 60. Yates PJ, Hazen R, St Clair M, Boone L, Tisdale M, Elston RC. 2006. In vitro development of resistance to human immunodeficiency virus protease inhibitor GW640385. Antimicrob. Agents Chemother. 50:1092– 95 61. Cihlar T, He GX, Liu X, Chen JM, Hatada M, et al. 2006. Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol. 363:635–47 www.annualreviews.org • New Antivirals and Drug Resistance 115 ARI 29 April 2009 10:11 62. Callebaut C, Stray KM, Tsai L, Xu L, He G-X, et al. 2007. GS-8374, a novel phosphonate HIV protease inhibitor with potent in vitro antiretroviral activity, low metabolic toxicity, and favorable resistance profile. Antivir. Res. 74:A27 63. Maguire MF, Guinea R, Griffin P, Macmanus S, Elston RC, et al. 2002. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 76:7398–406 64. De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, et al. 2005. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 49:2314–21 65. Nijhuis M, van Maarseveen NM, Lastere S, Schipper P, Coakley E, et al. 2007. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 4:e36 66. Callebaut C, Stray KM, Tsai L, Xu L, Lee W, Cihlar T. 2007. In vitro HIV-1 resistance selection to GS8374, a novel phosphonate protease inhibitor: comparison with Lopinavir, Atazanavir and Darunavir. Antivir. Ther. 12:S18 67. Wiley DC, Wilson IA, Skehel JJ. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–78 68. Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, et al. 1985. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317:145–53 69. Gerhard W, Yewdell J, Frankel ME, Webster R. 1981. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290:713–17 70. Knossow M, Daniels RS, Douglas AR, Skehel JJ, Wiley DC. 1984. Three-dimensional structure of an antigenic mutant of the influenza virus haemagglutinin. Nature 311:678–80 71. Varghese JN, Webster RG, Laver WG, Colman PM. 1988. Structure of an escape mutant of glycoprotein N2 neuraminidase of influenza virus A/Tokyo/3/67 at 3 A. J. Mol. Biol. 200:201–3 72. Tulip WR, Varghese JN, Webster RG, Laver WG, Colman PM. 1992. Crystal structures of two mutant neuraminidase-antibody complexes with amino acid substitutions in the interface. J. Mol. Biol. 227:149– 59 73. Malby RL, Tulip WR, Harley VR, McKimm-Breschkin JL, Laver WG, et al. 1994. The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 2:733–46 74. Dall’Acqua W, Goldman ER, Eisenstein E, Mariuzza RA. 1996. A mutational analysis of the binding of two different proteins to the same antibody. Biochemistry 35:9667–76 75. Colman PM, Laver WG, Varghese JN, Baker AT, Tulloch PA, et al. 1987. Three-dimensional structure of a complex of antibody with influenza virus neuraminidase. Nature 326:358–63 76. Kwong PD. 2005. Human immunodeficiency virus: refolding the envelope. Nature 433:815–16 77. Colman PM. 1997. Virus versus antibody. Structure 5:591–93 78. Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, et al. 2008. The global circulation of seasonal influenza A (H3N2) viruses. Science 320:340–46 79. Salzberg S. 2008. The contents of the syringe. Nature 454:160–61 80. Binley JM, Wrin T, Korber B, Zwick MB, Wang M, et al. 2004. Comprehensive cross-clade neutralization analysis of a panel of antihuman immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78:13232–52 81. Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, et al. 2004. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 5:233–36 82. Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, et al. 2001. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 293:1155–59 83. Saphire EO, Montero M, Menendez A, van Houten NE, Irving MB, et al. 2007. Structure of a highaffinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J. Mol. Biol. 369:696–709 84. Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, et al. 2003. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300:2065–71 Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 116 Colman Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 ARI 29 April 2009 10:11 85. Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, et al. 2004. Structure and mechanistic analysis of the antihuman immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 78:10724–37 86. Pai EF, Klein MH, Chong P, Pedyczak A. 2000. Int. Patent No. WO-00/61618 87. Zwick MB, Komori HK, Stanfield RL, Church S, Wang M, et al. 2004. The long third complementaritydetermining region of the heavy chain is important in the activity of the broadly neutralizing antihuman immunodeficiency virus type 1 antibody 2F5. J. Virol. 78:3155–61 88. Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, et al. 2005. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22:163–73 89. Cardoso RM, Brunel FM, Ferguson S, Zwick M, Burton DR, et al. 2007. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J. Mol. Biol. 365:1533–44 90. Walker BD, Burton DR. 2008. Toward an AIDS vaccine. Science 320:760–64 91. Fauci AS, Johnston MI, Dieffenbach CW, Burton DR, Hammer SM, et al. 2008. HIV vaccine research: the way forward. Science 321:530–32 92. Zhao X, Chen FP, Megaw AG, Sullender WM. 2004. Variable resistance to palivizumab in cotton rats by respiratory syncytial virus mutants. J. Infect. Dis. 190:1941–46 93. Smith BJ, Lawrence MC, Colman PM. 2002. Modelling the structure of the fusion protein from human respiratory syncytial virus. Protein Eng. 15:365–71 94. Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, et al. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure 9:255–66 95. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 102:9288–93 96. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 97. Zhao X, Sullender WM. 2005. In vivo selection of respiratory syncytial viruses resistant to palivizumab. J. Virol. 79:3962–68 98. Zhao X, Liu E, Chen FP, Sullender WM. 2006. In vitro and in vivo fitness of respiratory syncytial virus monoclonal antibody escape mutants. J. Virol. 80:11651–57 99. Simon A, Ammann RA, Wilkesmann A, Eis-Hubinger AM, Schildgen O, et al. 2007. Respiratory syncytial virus infection in 406 hospitalized premature infants: results from a prospective German multicentre database. Eur. J. Pediatr. 166:1273–83 99a. Keck ZY, Machida K, Lai MM, Ball JK, Patel AH, Foung SK. 2008. Therapeutic control of hepatitis C virus: the role of neutralizing antibodies. Curr. Top. Microbiol. Immunol. 317:1–38 100. Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–69 101. Lamb RA, Jardetzky TS. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427–36 102. Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. 1992. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 89:10537–41 103. Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770–74 104. Jiang S, Lin K, Strick N, Neurath AR. 1993. HIV-1 inhibition by a peptide. Nature 365:113 105. Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–73 106. Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–30 107. Lu J, Deeks SG, Hoh R, Beatty G, Kuritzkes BA, et al. 2006. Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: results of a clonal analysis. J. Acquir. Immune Defic. Syndr. 43:60–64 www.annualreviews.org • New Antivirals and Drug Resistance 117 ARI 29 April 2009 10:11 108. Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 72:986–93 109. Labrosse B, Morand-Joubert L, Goubard A, Rochas S, Labernardiere JL, et al. 2006. Role of the envelope genetic context in the development of enfuvirtide resistance in human immunodeficiency virus type 1infected patients. J. Virol. 80:8807–19 110. Mink M, Mosier SM, Janumpalli S, Davison D, Jin L, et al. 2005. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J. Virol. 79:12447–54 111. Liu S, Jing W, Cheung B, Lu H, Sun J, et al. 2007. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J. Biol. Chem. 282:9612–20 112. Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. 1999. Inhibiting HIV-1 entry: discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–15 113. Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 104:16828–33 114. Chinnadurai R, Rajan D, Munch J, Kirchhoff F. 2007. Human immunodeficiency virus type 1 variants resistant to first- and second-version fusion inhibitors and cytopathic in ex vivo human lymphoid tissue. J. Virol. 81:6563–72 115. Eron JJ, Gulick RM, Bartlett JA, Merigan T, Arduino R, et al. 2004. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J. Infect. Dis. 189:1075–83 116. Melby T, Demasi R, Cammack N, Miralles GD, Greenberg ML. 2007. Evolution of genotypic and phenotypic resistance during chronic treatment with the fusion inhibitor T-1249. AIDS Res. Hum. Retrovir. 23:1366–73 117. Martin-Carbonero L. 2004. Discontinuation of the clinical development of fusion inhibitor T-1249. AIDS Rev. 6:61 118. Dwyer JJ, Wilson KL, Martin K, Seedorff JE, Hasan A, et al. 2008. Design of an engineered N-terminal HIV-1 gp41 trimer with enhanced stability and potency. Protein Sci. 17:633–43 119. Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, et al. 2008. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J. Virol. 82:6678–88 120. He Y, Xiao Y, Song H, Liang Q, Ju D, et al. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 283:11126–34 121. Root MJ, Kay MS, Kim PS. 2001. Protein design of an HIV-1 entry inhibitor. Science 291:884–88 122. Luftig MA, Mattu M, Di Giovine P, Geleziunas R, Hrin R, et al. 2006. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat. Struct. Mol. Biol. 13:740–47 123. Steger HK, Root MJ. 2006. Kinetic dependence to HIV-1 entry inhibition. J. Biol. Chem. 281:25813–21 124. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–42 125. DeLano WL. 2007. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Sci. http:// www.pymol.org/ Annu. Rev. Biochem. 2009.78:95-118. Downloaded from arjournals.annualreviews.org by University of Texas - Austin on 10/05/09. For personal use only. ANRV378-BI78-05 118 Colman