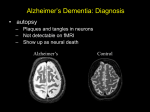
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
doi:10.1093/brain/awh356 Brain (2005), 128, 752–772 Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation Bradley F. Boeve,1,5 Ivo W. Tremont-Lukats,6 Andrew J. Waclawik,7 Jill R. Murrell,9 Bruce Hermann,7 Clifford R. Jack Jr,2,5 Maria M. Shiung,2 Glenn E. Smith,3,5 Anil R. Nair,1,5 Noralane Lindor,4 Vinaya Koppikar8 and Bernardino Ghetti9 Departments of 1Neurology, 2Diagnostic Radiology, Psychiatry and Psychology and 4Medical Genetics, Mayo Clinic College of Medicine, Rochester, Minnesota, MN, 5 Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation, 6Department of Neurology, Medical University of South Carolina, Charlston, SC, 7Department of Neurology, University of Wisconsin, Madison, 8Monroe Clinic, Monroe, WI and 9Indiana Alzheimer Disease Center and Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN, USA 3 Correspondence to: Bradley F. Boeve, MD, Division of Behavioral Neurology, Department of Neurology, Mayo Clinic, 200 1st Street SW, Rochester, MN 55905, USA E-mail: [email protected] Summary The background to this study began with the reporting of two Japanese kindreds with the S305N tau mutation. Although the pathological findings in the autopsied cases were well characterized, only limited ante-mortem data were presented. In this study, longitudinal characterization was carried out in two siblings of European ancestry found to have frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) through comprehensive neurobehavioural examinations and other scales at approximate 6-month intervals. Scales included the Mini-Mental State Examination, Short Test of Mental Status, modified motor subtest of the Unified Parkinson’s Disease Rating Scale, detailed neuropsychological testing, and the Neuropsychiatric Inventory. Changes in whole-brain volume and ventricular volume were measured from serial MRI studies. All members of the kindred underwent molecular genetic analyses to elucidate the mechanism of inheritance. The missense mutation in tau, S305N, was detected in the proband (onset age 30), who has undergone serial evaluations for almost 4 years. Her older sister (onset age 36) was subsequently found to have the same mutation, and has undergone serial evaluations for 2 years. This mutation is absent in both parents and the only other sibling, and non-paternity was excluded by additional analyses. The siblings have exhibited cognitive and behavioural features typical of FTDP-17, which have proved challenging to manage despite aggressive pharmacological and behavioural therapies. The proband’s sister has demonstrated an atypical profile of impairment on neuropsychological testing. Both siblings have developed striking atrophy of the anterior part of temporal lobes and moderate atrophy of the dorsolateral and orbitofrontal cortical regions, which in both cases is relatively symmetrical. The annualized changes in whole-brain volume and ventricular volume, respectively, were 35.2 ml/year (3.23% decrease per year) and +20.75 ml/year (16.93% increase per year) for the proband, and 30.75 ml/year (2.77% decrease per year) and +5.01 ml/ year (3.11% increase per year) for the proband’s sister. In conclusion, the mutation in these siblings may have arisen during oogenesis in the mother and probably represents germline mosaicism. Although both patients have exhibited the typical cognitive and behavioural features of FTDP-17, one patient is exhibiting an atypical neuropsychological profile. Also, despite a similar topographic pattern of progressive atrophy on MRI, the rates of change in whole-brain volume and ventricular volume between the two patients are quite different. These findings have implications for future drug trial development in FTDP-17 and the sporadic tauopathies. # The Author (2005). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: [email protected] FTDP-17 associated with the S305N tau mutation 753 Keywords: frontotemporal dementia; tau; MAPT; neuropsychology; magnetic resonance imaging Abbreviations: Ab42 = b-amyloid 1–42; CDR = Clinical Dementia Rating; COWAT = Controlled Oral Word Association Test; CSF = cerebrospinal fluid; DRS = Mattis Dementia Rating Scale; FLAIR = fluid attenuation inversion recovery; FTD = frontotemporal dementia; FTDP-17 = frontotemporal dementia and parkinsonism linked to chromosome 17; GDS = Global Deterioration Scale; MAPT = gene encoding tau (microtubule associated protein tau); MLAE = Multilingual Aphasia Examination; MMSE = Mini-Mental State Examination; NPI = Neuropsychiatric Inventory; Rey-O = Rey–Osterrieth complex figure; ROIL = Record of Independent Living; STMS = Short Test of Mental Status; Stroop = Stroop Neurological Screening Test; UPDRS = Unified Parkinson’s Disease Rating Scale; VV = ventricular volume; WAIS-III = Wechsler Adult Intelligence Scale—Third Edition; WBV = whole-brain volume; WCST = Wisconsin Card Sorting Test Received April 16, 2004. Revised August 1, 2004. Accepted October 26, 2004. Advance Access publication December 22, 2004 Introduction Frontotemporal dementia (FTD) is a syndrome characterized by prominent behavioural and/or dysexecutive features (Neary et al., 1998; McKhann et al., 2001; Miller et al., 2003). Anterograde memory functioning and visuospatial skills are typically preserved early in the course of the illness. Frontal and/or temporal abnormalities are demonstrated on structural and functional neuroimaging studies. FTD occurs in familial and sporadic forms; frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) is one type characterized by mutations in MAPT (gene encoding tau), which are inherited in an autosomal dominant pattern. Its neuropathological hallmark is an abundance of hyperphosphorylated tau protein and degeneration of neurons and glia (Munoz et al., 2003). Extensive research in the past 6 years has revealed well over 30 different mutations in tau (Reed et al., 2001; Bird et al., 2003; Ghetti et al., 2003). While the genetic, biochemical and neuropathological features of affected individuals have been well characterized, there is remarkably little longitudinal data on the clinical, neuropsychological and radiological features. With the hope that agents potentially useful in the treatment of FTDP-17 and the sporadic tauopathies may some day be identified or developed, longitudinal antemortem data will be necessary for designing clinical trials. Two Japanese kindreds with the S305N tau mutation have been reported (Iijima et al., 1999; Kobayashi et al., 1999, 2002, 2003, 2004). Although the pathological findings in the autopsied cases were well characterized, only limited ante-mortem data were presented. No FTDP-17 patients with the S305N mutation have been identified outside of Japan to our knowledge. In this paper, we present genetic and detailed longitudinal clinical, neuropsychological, neuropsychiatric and radiological data on two young Caucasian female siblings with FTDP-17 associated with the S305N tau mutation. Methods and design The two patients are enrolled in the Mayo Foundation Institutional Review Board-approved Alzheimer’s Disease Research Center at Mayo Clinic, Rochester, MN. Genetic analyses, cerebrospinal fluid (CSF) analyses for tau and b-amyloid 1–42 (Ab42) and serial neuropsychological assessments and MRI scans were performed after the patients and family provided appropriate informed consent. Both patients underwent a standardized battery of clinical, functional, neuropsychological, neuropsychiatric and radiological studies at approximately 6-month intervals. Clinical evaluations All comprehensive neurobehavioural clinical data (Members of the Department of Neurology, 1998) were reviewed and summarized. The modified motor subtest of the Unified Parkinson’s Disease Rating Scale (UPDRS) was used to assess the degree of parkinsonism (range 0–44); increasing values reflect greater degrees of parkinsonism (Fahn et al., 1987). Functional assessments All data on the Clinical Dementia Rating (CDR) scale (Morris, 1993), Global Deterioration Scale (GDS) (Reisberg et al., 1988) and Record of Independent Living (ROIL) parts A and B (Weintraub, 1986) were tabulated and analysed. Neuropsychological assessments Testing included assessments of screening and global cognitive functioning [Mini-Mental State Examination (MMSE) (Folstein et al., 1975), Short Test of Mental Status (STMS) (Kokmen et al., 1991; Tang-Wai et al., 2003), Mattis Dementia Rating Scale (DRS) (Mattis, 1988), Neurobehavioral Cognitive Status Examination, 1988], academic achievement [Wide Range Achievement Test—3: Reading (WRAT-3) (Wilkinson, 1993)], intellectual functioning [Kaufman Brief Intelligence Test (Kaufman and Kaufman, 1990), Wechsler Adult Intelligence Scale—Third Edition (WAIS-III) (Wechsler, 1997a)], attention/executive functioning [Trail Making Test (Reitan, 1958), Stroop Neurological Screening Test (Trenerry et al., 1989), Digit Span of the WAIS-III, Digit Symbol-Coding of the WAIS-III, Symbol Search of the WAIS-III and Similarities of the WAIS-III, and Wisconsin Card Sorting Test (WCST) (Heaton, 1981)], language functioning [Boston Naming Test (Kaplan et al., 1978), Controlled Oral Word Association Test (COWAT) (Benton and Hamsher, 1978), Multilingual Aphasia Examination (MLAE) Naming, Token, and Sentence Repetition (Benton and Hamsher, 1978), and category/semantic fluency (animals, fruit, vegetables)], learning and memory [Wechsler Memory Scale—Third Edition (WMS-III) (Wechsler, 1997b), Rey-Auditory Verbal Learning Test (Rey, 1964), and Free and Cued Selective Reminding Test 754 B. F. Boeve et al. (Free and Cued Selective Reminding Test) (Buschke, 1984; Grober and Buschke, 1987)], and visuoconstructive and visuospatial skills [Rey–Osterreith complex figure (Rey-O) (Osterrieth, 1944; Rey, 1941), Judgement of Line Orientation (Benton et al., 1983), Facial Recognition Test (Benton et al., 1983) and Visual Form Discrimination (Visual Form Discrimination) (Benton et al., 1983)]. All scores were converted to Z scores (mean of zero and standard deviation of 1) based on comparison with appropriate norms. Neuropsychiatric assessments Neuropsychiatric Inventory (NPI) (Cummings et al., 1994) data were provided by the proband’s husband (caregiver of the proband) and mother (caregiver of the proband’s sister). Neuroimaging examinations MRI was performed using a GE scanner at 1.5 tesla, and images of the brain were obtained in the sagittal (T1-weighted), axial [protondensity, T2-weighted and fluid attenuation inversion recovery (FLAIR)] and coronal (T1-weighted) planes. Changes in wholebrain volume (WBV) and ventricular volume (VV) were measured from serial MRI studies using the boundary shift integral method as described by Gunter and colleagues (Gunter et al., 2003). Genetic analyses MAPT (the gene encoding tau) analysis was performed as previously described (Spillantini et al., 1998). For the proband, exons 1–5, 7 and 9–13 were amplified and directly sequenced. Primers from the intronic sequences surrounding the exons were used so that the entire exon sequence and the splice signals could be analysed. Standard amplification reactions were done with 50 ng of genomic DNA, and the amplified products were then gel-purified. Asymmetrical amplification using the DTCS Quick Start Kit (Beckman Coulter, Fullerton, CA, USA) was performed. The amplified products were precipitated and resuspended in sample loading solution and loaded onto a CEQ 200XL DNA Analysis System (Beckman Coulter). The sequences were compared with those of normal controls and to the published MAPT sequence. Once a pathogenic tau mutation was identified in the proband, her family underwent genetic counselling but no additional analyses were performed until after the proband’s sister developed symptoms. All members of this kindred eventually underwent tau mutation analyses using both direct sequencing and BsrDI restriction enzyme analyses after appropriate genetic counselling. For enzyme digestion analyses, the primers 50 -GACTGCCTCTGCCAAGTCCG-30 and 50 GGATCTGGCTGCGACCTCTG-30 were used to amplify the MAPT exon 10. The 383 bp amplification product was then digested with BsrDI, which recognizes the mutant allele and resulted in two bands of sizes 252 and 131 bp. To determine if the mother had any sign of the mutant allele and to verify that the polymorphism (adenine at position 47) was indeed on the normal allele, amplified exon 10 products from the proband and the proband’s affected sister, father and mother were cloned into pBluescript (Stratagene, La Jolla, CA, USA). Clones were analysed by restriction enzyme digestions with BsrDI (New England Biolabs, Beverly, MA, USA) and AciI, which was used to determine the polymorphism (C/A) 47 bp upstream of the start of exon 10. A few clones were sequenced as described above. To determine if non-paternity might explain the genetic results in this kindred, a standard paternity test was run in all five subjects, which included nine polymorphic markers: D3S1358, tetranucleotide repeats in the von Willebrand (vWA) gene and the fibrinogen A a polypeptide (FGA) gene, D8S1179, D21S11, D18S51, D5S818, D13S317, and D7S820. Markers were analysed using the AmpFLSTR Profiler Plus PCR Amplification kit (Applied Biosystems) on an ABI Prism 310 Genetic Analyzer (Applied Biosystems). Plasma and CSF tau and b-amyloid analyses The CSF levels of tau and Ab42 in the proband were determined using previously described methods by Athena Diagnostics. Results Pedigree The pedigree is shown in Fig. 1. The maternal lineage is of Bulgarian, English, Dutch and Swedish ancestry. The paternal lineage is of Swedish and Norwegian ancestry. Case reports Case II.2 The proband is a right-handed Caucasian woman with 14 years of formal education who began exhibiting cognitive impairment and behavioural changes at age 30. Over the subsequent few months, she lost the ability to adequately make decisions and execute instructions at work, resulting in her dismissal. Her social behaviour changed such that she altered her hair and dressing style to look similar to her teenage daughter, her expressions of affection to her family disappeared, and she became easily irascible and started to withdraw socially. She drove erratically and her driving privileges were revoked. At age 32 she became obsessed with financial matters, was apathetic for upkeep of the home, was intermittently aggressive towards her son, and exhibited manic behaviour. She ate increasing amounts of candy. By age 33 she was dependent on her husband for most activities of daily living. She was evaluated by a psychiatrist and commenced on risperidone, sertraline and buspirone, I. c=63 c=60 2 1 II. o=36 c=39 o=30 c=36 1 c=34 2 3 Fig. 1 Pedigree. The shaded circles represent the two affected individuals with FTD and the arrow indicates the proband. The ages of onset (o = XX) and current ages (c = XX) are also shown. FTDP-17 associated with the S305N tau mutation which improved her aggressive behaviour. Valproic acid was subsequently added, which improved her manic symptomatology. The past medical, family and social history of this young woman at presentation was only notable for a sister with cerebral palsy. Both parents were in their 50s and did not have cognitive or behavioural symptoms. Her only other sibling (case II.1) was not symptomatic at the time of her presentation. Initial neurological examination revealed a well-groomed, adequately dressed young woman who avoided eye contact. Her face was hypomimic, impassive throughout the encounter, looking repeatedly at her watch. She scored 29/30 on the MMSE, missing one point on recall. She had difficulty naming the last five presidents of the USA. The remainder of her examination was entirely normal. At the initial neuropsychological testing (Table 1), general intellectual functioning appeared to have fallen from an estimated premorbid level of average to low average level. Expressive language skills were already moderately impaired but receptive skills remained intact. Visuospatial/visuoconstructive functioning was preserved. Immediate and delayed recall of both verbal and non-verbal information was mildly impaired. The patient’s perseveration during problem-solving was exceptional. Overall neurocognitive function was mildly impaired. Numerous studies on blood and CSF failed to reveal findings suggesting an infectious, autoimmune/inflammatory, toxic, metabolic, paraneoplastic or mitochondrial process. Awake and sleep electroencephalography was normal. An initial MRI of the brain with and without contrast revealed regional volume loss affecting the frontal and temporal lobes (not shown). Tau and Ab peptide concentrations in CSF were 478 pg/ml (normally <420 pg/ml) and 521 pg/ml (normally <1240 pg/ml), respectively. Despite the lack of dementia in her immediate family, she and her family underwent genetic counselling and genetic testing. Apolipoprotein (ApoE) genotyping was negative for the ApoE e4 allele. MAPT analysis was then performed because of her young age and the mildly elevated CSF tau level. Sequencing of exon 10 of MAPT revealed a G to A substitution at the second base of codon 305, resulting in a serine to asparagine amino acid change (S305N). She was diagnosed with FTDP-17 due to the S305N tau mutation, and donepezil was added to her medical regimen and her alertness and communicability appeared to improve. Examination 4 months later (age 34) revealed abulia and bradyphrenia. On the STMS she scored 25/38. She recalled none of four words on delay. She copied the Rey–Osterrieth complex figure well. She had a mild hypokinetic dysarthria, mild rigidity in the axial musculature and right upper extremity with minimal rigidity elsewhere, moderately masked facies, mildly stooped posture, moderately decreased arm swing bilaterally, mild postural but not rest tremor, mild diffuse bradykinesia, mildly decreased upgaze with slight impairment of saccadic eye movements on pursuit testing, and diffuse hyper-reflexia without pathological pyramidal 755 Table 1 Neuropsychometric performance in the proband (case II.2) Age (years, months) 33, 3 Global functioning/screening Mini-Mental State Examination 29 (raw) Short Test of Mental Status (raw) Academic achievement Wide Range Achievement Test—3: Reading 0 Intellectual functioning Kaufman Brief Intelligence Test Vocabulary 1.0 Matrices 1.2 Composite IQ 1.2 Attention/concentration Trail-Making Test A (s) 0.6 Trail-Making Test B (s) 0.1 Stroop, word 1.1 Stroop, colour 0.1 Stroop, colour-word 1.0 Language functioning COWAT 1.9 MLAE naming 2.0 MLAE token 0.8 MLAE sentence repetition 0.4 Planning, reasoning, problem-solving Wisconsin Card Sorting Test Categories 2.0 Perseverative responses 3.0 Failure to maintain set 0 Learning and memory Wechsler Memory Scale—III Auditory immediate 1.1 Visual immediate 1.6 Immediate memory 1.7 Auditory delayed 1.5 Visual delayed 2.1 Auditory recognition delayed 0.6 General memory 1.8 Working memory 0 Visuospatial functioning Rey–Osterrieth complex figure copy Facial recognition test 0.2 Judgement of line orientation 2.3 Visual form discrimination 1.2 33, 7 33, 9 34, 2 25 25 23 0 2.0 2.5 2.5 1.1 1.6 2.5 1.3 2.9 3.1 3.6 3.2 2.7 2.7 3.6 0.6 2.6 0.4 2.0 0.3 Unless otherwise noted, values reflect Z scores (mean = 0, SD = 1) based on appropriate norms. tract signs. Glabellar and palmomental reflexes were present. Repeat neuropsychological testing (performed 6 months following the initial assessment) (Table 1) demonstrated that general intellectual function had fallen to the severely impaired range. Impairment had appeared in all assessed cognitive domains. Memory function was profoundly impaired. Receptive language deficits had joined the expressive deficits. Mild alternating attention problems had appeared. Of the abilities assessed, only spatial judgement skills remained 756 B. F. Boeve et al. Table 2 Responses on the Neuropsychiatric Inventory and scores on clinical and functional scales for the proband (case II.2) Age (yrs/mo) 33, 7 Feature Freq Delusions Hallucinations Agitation Depression/dysphoria Anxiety Euphoria/elation Apathy/indifference Disinhibition Irritability/lability Aberrant motor behaviour Night-time behaviour Appetite/eating change CDR GDS ROIL Part A ROIL Part A UPDRS Medications 0 0 3 0 3 0 4 1 0 3 0 3 Sev 34, 2 Dis 0 0 0 0 2 2 0 0 1 1 0 0 2 2 1 1 0 0 2 2 0 0 2 2 1 4 30 8 25 vitamin E buspirone donepezil risperidone sertraline Age (yrs/mo) Freq 0 0 3 0 3 0 4 2 3 4 4 0 Sev 34, 8 Dis Freq 0 0 0 0 1 3 0 0 2 3 0 0 3 3 1 1 2 3 2 3 2 4 0 0 2 5 35 9 16 vitamin E buspirone donepezil valproic acid memantine quetiapine risperidone trazodone ibuprofen lorazepam 0 0 3 0 4 4 4 1 2 4 4 4 36, 3 Feature Freq Delusions Hallucinations Agitation Depression/dysphoria Anxiety Euphoria/elation Apathy/indifference Disinhibition Irritability/lability Aberrant motor behaviour Night-time behaviour Appetite/eating change CDR GDS ROIL Part A ROIL Part B UPDRS Medications 0 0 3 0 0 0 4 3 0 4 0 3 Sev 0 0 1 0 0 0 2 1 0 1 0 2 3 6 58 11 9 vitamine E buspirone donepezil valproic acid memantine quetiapine gabapentin mirtazepine ibuprofen Sev 35, 2 Dis 0 0 0 0 1 2 0 0 2 3 1 1 2 1 1 1 2 3 2 4 3 4 2 2 2 6 38 8 4 vitamin E buspirone donepezil valproic acid memantine quetiapine trazodone gabapentin ibuprofen Freq 0 0 3 0 0 0 4 3 3 4 0 4 Sev 35, 7 Dis 0 0 0 0 1 3 0 0 0 0 0 0 1 2 1 1 2 2 2 2 0 0 2 2 2 6 40 8 3 vitamin E buspirone donepezil valproic acid memantine quetiapine mirtazapine gabapentin ibuprofen 36, 8 Dis Freq 0 0 0 0 0 0 0 2 0 0 0 2 0 0 4 0 3 0 4 0 0 4 0 0 Sev 0 0 1 0 2 0 3 0 0 3 0 0 3 6 60 10 10 vitamin E buspirone donepezil valproic acid memantine quetiapine ibuprofen Freq 0 0 4 0 3 0 4 3 0 4 3 4 Sev Dis 0 0 0 0 2 3 0 0 2 0 0 0 3 0 1 0 0 0 1 0 2 3 3 4 3 6 56 11 14 vitamin E buspirone donepezil valproic acid memantine quetiapine mirtazapine gabapentin ibuprofen 37, 2 Dis Freq 0 0 1 0 2 0 2 0 0 0 0 0 0 0 4 0 0 0 4 3 0 4 3 0 Sev 0 0 1 0 0 0 2 1 0 1 2 0 3 6 63 11 5 vitamin E buspirone donepezil valproic acid memantine quetiapine ibuprofen CDR = Clinical Dementia Rating scale, Dis = distress, Freq = frequency, GDS = Global Deterioration Scale, ROIL = Record of Independent Living, Sev = severity, UPDRS = Unified Parkinson’s Disease Rating Scale (modified). Dis 0 0 0 0 0 0 0 0 0 0 0 0 FTDP-17 associated with the S305N tau mutation within normal limits (though decline in performance was present here also). Neurocognitive level was rated as moderately impaired. Her husband’s responses to questioning on the NPI are shown in Table 2. MRI (Figs 2A and 3A) showed moderate atrophy involving the amygdala, hippocampi and frontotemporal cortex. A B C Fig. 2 Continued on next page. 757 She was commenced on vitamin E 2000 IU/day for its putative antioxidant effects as well as ibuprofen for its anti-inflammatory properties. Over the subsequent 4 months, severe insomnia and wandering developed which initially responded well to trazodone. Memantine was added 2 months later. Insomnia, wandering and intermittent aggressive 758 B. F. Boeve et al. D E F Fig. 2 Coronal T1-weighted MRIs of case II.2 (proband) at ages 33 years 7 months (A), 34 years 2 months (B), 34 years 8 months (C), 35 years 7 months (D), 36 years 3 months (E) and 37 years 2 months (F). Atrophy is initially most prominent in the amygdalae (right > left), anterior temporal cortices and dorsolateral prefrontal cortices. The left lateral ventricle is slightly larger than the right. Over time the ventricles enlarge and the atrophy progresses, with eventual profound atrophy in the amygdalae, hippocampi and frontotemporal cortices. Note that the findings are largely symmetrical, particularly as the illness progresses, with the mesial and inferior temporal structures more atrophic than the middle and superior temporal gyri. The orbitofrontal cortex is minimally atrophic early (A–C) and severely atrophic late in the course (D–F). FTDP-17 associated with the S305N tau mutation behaviour escalated and sertraline and risperidone were tapered off and quetiapine was added. Her ability to perform some activities of daily living improved, as did her neuropsychiatric features, but insomnia and wandering remained problematic. A B C Fig. 3 Continued on next page. 759 She was re-evaluated 7 months following the previous examination. She was alert, had fair recall of recent events in the news, repeatedly expressed her desire to leave the examining room, was dysnomic with normal fluency, repetition and comprehension, and had delayed responses 760 B. F. Boeve et al. D E Fig. 3 Axial FLAIR MRIs of case II.2 (proband) at ages 33 years 7 months (A), 34 years 2 months (B), 34 years 8 months (C), 36 years 3 months (D) and 37 years 2 months (E); movement artefact obscured axial images at age 35 years 7 months, so these images are not shown. Note the progressive anteromesial temporal and frontal atrophy with associated ventricular dilatation, with the left lateral ventricle slightly larger than the right early in the course. No significant signal changes have developed in the affected frontotemporal structures, and only mild atrophy has evolved in the parietal cortices. to questions. She scored 23 out of 38 on the STMS with errors in calculations, abstraction and recall. Her copy of the Rey–Osterrieth complex figure was again almost perfect. Parkinsonism was similar to that noted on the prior examination, and frontal release signs were now absent. Her husband’s responses to questioning on the NPI are shown in Table 2. MRI (Figs 2B and 3B) showed significant progressive atrophy involving the amygdala, hippocampi and frontotemporal cortex. Atrophy was particularly prominent along the interhemispheric fissure and in the inferior and middle temporal gyri. Quetiapine was titrated upwards to further manage insomnia and intermittent agitation. She was re-evaluated 6 months later. As insomnia, manic as well as obsessive–compulsive features had escalated, quetiapine had been increased to 500 mg daily, valproic acid was increased to 2000 mg daily, and gabapentin was added, yet these neuropsychiatric features continued. Her husband was averaging 4–5 h of sleep nightly. He was pleased to see expressions of emotion and affection and intermittent periods of remarkable lucidity following these pharmacological manipulations. Her parkinsonian features had dissipated. She scored 10 out of 30 on the MMSE. She did not answer most questions on the STMS. She abruptly left the office five times to use the bathroom. Language was largely fluent with poor comprehension and repetition. There was mildly reduced upgaze on eye movement testing, subtle rigidity in the left upper extremity, very mild postural tremor, moderately reduced facial animation, but no other parkinsonian signs. Frontal release signs were again absent. Responses on the NPI are shown in Table 2. MRI (Figs 2C and 3C) showed slightly more anterior temporal atrophy and slight ventricular enlargement. Her trazodone and quetiapine were increased, and she continues under the diligent care of her supportive family. Longitudinal data over the subsequent 2 years are shown in Tables 1 and 2 and Figs 2D–F and 3D and E. Thus far, over almost FTDP-17 associated with the S305N tau mutation 4 years of longitudinal evaluations she has not developed apraxia of speech, non-verbal oral apraxia, limb apraxia, alien limb phenomenon, dystonia, chorea, ataxia, myoclonus, downgaze palsy, fasciculations, pyramidal tract signs or muscular atrophy. Case II.1 The proband’s older sister (henceforth referred to as ‘the sister’) began exhibiting child-like behaviour at age 36. Although considered the most outgoing and gregarious of the family, her behaviour was considered beyond the norm for her. She tended to laugh and giggle at most topics of conversation, did not seem to appreciate the levity of some inappropriate behaviour of her 12-year-old daughter, and tended to interact with her daughter more as a sister or good friend rather than a mother. She rarely disciplined her daughter. The patient was divorced from her first husband and separated from her second husband, and her parents were the primary caregivers for the patient and only source of discipline for her daughter. She lost her job at age 37 and spent much of her time fishing or sunbathing. Her problem-solving skills and judgement had declined, but neither she nor her parents appreciated any significant problems in memory, language or visuospatial functioning. There were no symptoms of motor dysfunction. These behaviours led to genetic counselling and genetic testing, and the same S305N tau mutation was identified. Apolipoprotein E testing showed no e4 alleles. Shortly thereafter the sister underwent her first evaluation at Mayo Clinic Rochester. Initial neurological examination at age 37 revealed an alert, attentive young woman who provided a good history, and had excellent recall of recent events in the news. She scored 32 out of 38 on the Kokmen STMS, taking two trials to learn four items, missing two on calculations, two on abstraction and one on recall. There was no significant language disturbance evident in office testing. She tended to giggle after many of her statements. The remainder of her comprehensive neurological examination was notable for mild generalized hyper-reflexia, Hoffmann’s sign present bilaterally, slightly more evident on the left than right, and no significant rigidity except with contralateral limb movements, as tone increased significantly on the left, mildly so on the right. There were no other extrapyramidal signs and no gaze palsy, apraxia, myoclonus, etc., and no findings of motor neuron disease. Her neuropsychological test results are shown in Table 3; these results are largely within normal limits except for deficits in confrontation naming and recall of verbal material. Her mother’s responses on the NPI are shown in Table 4. Her MRI of the brain showed mild to moderate bilateral amygdala atrophy, as well as mild hippocampal atrophy, minimal atrophy along the falx, no significant atrophy elsewhere, and no significant vascular changes (Figs 4A and 5A). She was diagnosed with early FTDP-17, enrolled in the Mayo Alzheimer’s Disease Research Center, and was closely followed by her local psychiatrist. She was re-evaluated 6 months later. She had become more forgetful, and her very poor insight continued with 761 no appreciation of anything amiss. She reluctantly discontinued driving. Her personality/behavioural changes were most problematic, particularly disinhibition and sexually inappropriate gestures and behaviours. She was also becoming more euphoric, laughing at topics of conversation that were not overly funny. She tended to hoard meat and feed this to their dog rather than eat these items herself, and she greatly increased her intake of peanut butter. She was also compulsively buying items. Her medical regimen included memantine, valproic acid, venlafaxine and high-dose vitamin E. On examination she was alert, attentive, tended to giggle or laugh at times during the interview and examination. She scored 30 out of 38 on the STMS, missing one point each on orientation, attention, calculations, abstractions, taking two trials to learn one item, and missing three on recall. The remainder of her examination was essentially unchanged from 6 months previously. Neuropsychological testing (Table 3) showed little change except for mild worsening of expressive language problems. MRI of the brain (Figs 4B and 5B) showed slightly more atrophy along the falx, anterior temporal lobes, bilateral amygdala and bilateral hippocampi. The lateral ventricles were slightly larger. The patient, her parents and the patient’s daughter continued with care under the direction of local mental health professionals. The family had been impressed with the improvement in the proband with donepezil, and this was instituted in this patient with marginal benefit. Over the subsequent 6 months, she experienced some cognitive decline but still functioned reasonably well at home. She tended to watch game shows and play solitaire on her computer. She was enjoying sweets more, particularly cookies, cake, ice cream and certain types of cereal. She no longer cooked, and she continued to not discipline her daughter. Inappropriate behaviour towards her daughter had ceased, but her irritability towards her parents had escalated, with one occasion requiring her father to restrain her as she came at her mother with a knife. Quetiapine was commenced and within 2 weeks her irritability had greatly improved. On examination she scored 28 out of 38 on STMS, with no other significant changes in her general neurological examination. Neuropsychological findings were again minimally changed compared with the prior testing sessions (Table 3). MRI showed more atrophy in the amygdala, slightly more atrophy along the falx anteriorly, and the lateral ventricles were slightly more dilated (Figs 4C and 5C). Re-evaluation 6 months later revealed that most of her cognitive and behavioural issues were relatively stable, except for her feeding and shopping habits. She tended to eat icecream most of the day, resulting in a 20 pound increase in weight. She enjoyed going to stores and malls with her parents but occasionally her parents would find that she had shoplifted items when they were not looking. She continued to refrain from disciplining her daughter and would become very angry when her parents attempted to discipline her. On examination she scored 32 out of 38 on the STMS; her general examination was again notable for subtle parkinsonism, but absence of limb apraxia, dystonia, myoclonus, pyramidal tract signs, muscular weakness and atrophy, and frontal release signs. 762 B. F. Boeve et al. Table 3 Neuropsychometric performance in the sister (case II.1) Age (years, months) Global functioning/screening Mini-Mental State Examination (raw) Short Test of Mental Status (raw) Mattis Dementia Rating Scale (raw) Attention Initiation and perserveration Construction Conceptual Memory Total Academic achievement Wide Range Achievement Test—3 Reading Intellectual functioning Wechsler Adult Intelligence Scale—III Verbal IQ Performance IQ Full-scale IQ Verbal comprehension Perceptual organization Working memory Processing speed Attention/executive functioning Trail–Making Test A (s) Trail–Making Test B (s) Digit span of WAIS-III Digit symbol-coding of WAIS-III Symbol search of WAIS-III Similarities of WAIS-III Language functioning Boston Naming Test, long COWAT Category fluency (raw) MLAE token Learning and memory Wechsler Memory Scale–Revised Verbal Memory Index Logical memory Immediate Delayed Retention (%) Visual reproduction Immediate Delayed Retention (%) Auditory Verbal Learning Test Trial 5 Delay Delayed recall percent retention Visuospatial functioning Picture completion of WAIS-III Block design of WAIS-III Rey–Osterrieth complex figure copy Visual form discrimination 37, 3 37, 9 38, 3 38, 9 29 32 28 30 27 28 27 32 37 37 6 34 25 139 34 37 6 35 22 134 31 37 6 37 21 132 35 37 6 34 22 134 0.3 0.6 0.1 0.4 0.8 0 0.5 1.3 0 0.1 0.4 1.2 0 0.7 1.3 0 0.5 0 1.5 0.19 0.9 1.7 0 0.9 0.5 1.7 1.18 1.5 1.6 0.8 1.7 0.9 0.1 0.7 0 0.3 0.3 1.0 0.3 0.1 0.6 0 0 1.3 0.6 0.1 0.3 0.3 0.3 2.0 1.6 0.7 1.3 1.3 0.3 1.3 2.8 0.1 46 0.8 3.3 0.5 33 0.2 3.1 0 38 0.9 3.3 0.4 36 1.3 0.9 0.9 1.3 1.7 0.7 1.0 52 0.7 0.8 62 0.8 1.7 33 0.9 1.5 41 0.3 0.5 79 0.4 0.8 84 0.3 0.1 91 0.9 0.9 86 2.2 3.1 33 3.4 3.9 0 3.3 3.5 29 3.8 3.8 17 1.3 0 1.0 0.3 0.3 0 1.0 0.1 0.6 0.6 1.0 0.8 2.0 0.3 1.0 1.6 Unless otherwise noted, values reflect Z scores (mean = 0, SD = 1) based on appropriate norms. Her neuropsychological test scores remained stable in all spared areas and unchanged in the impaired naming and delayed recall areas (Table 3). MRI showed more atrophy in the mesial, inferior and lateral aspect of the anterior part of temporal lobes and anteriorly along the falx (Figs 4D and 5D). She was maintained on her pharmacological regimen and topiramate was added in hopes of improving her eating behaviour. She continues to reside with her parents. FTDP-17 associated with the S305N tau mutation 763 Table 4 Responses on the Neuropsychiatric Inventory and scores on clinical and functional scales for the sister (case II.1) Feature Age (years/months) 37, 3 Freq Delusions Hallucinations Agitation Depression/dysphoria Anxiety Euphoria/elation Apathy/indifference Disinhibition Irritability/lability Aberrant motor behaviour Night–time behaviour Appetite/eating change CDR GDS ROIL Part A ROIL Part B UPDRS Medications 0 0 0 0 0 4 0 4 1 0 0 0 Sev 0 0 0 0 0 1 0 1 2 0 0 0 0.5 1 4 0 3 Vitamin E 37, 9 Dis Freq 0 0 0 0 0 4 0 3 4 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 38, 3 Sev 0 0 0 0 0 2 0 0 0 0 0 0 1 3 6 1 2 Vitamin E Valproic acid Memantine Venlafaxine Dis Freq 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 1 0 3 4 3 1 0 0 4 Sev 0 0 0 2 0 2 2 2 1 0 0 2 0.5 4 17 2 2 Vitamin E Valproic acid Memantine Venlafaxine Quetiapine Donepezil 38, 9 Dis Freq 0 0 0 3 0 1 2 2 3 0 0 2 0 0 0 3 0 3 3 2 2 0 0 4 Sev 0 0 0 1 0 1 2 3 1 0 0 2 0.5 4 20 4 2 Vitamin E Valproic acid Memantine Venlafaxine Quetiapine Donepezil Dis 0 0 0 2 0 0 1 2 0 0 0 1 CDR = Clinical Dementia Rating scale; Dis = distress; Freq = frequency; GDS = Global Deterioration Scale; ROIL = Record of Independent Living; Sev = severity; UPDRS = Unified Parkinson’s Disease Rating Scale (modified). Analysis of genetic data All members of this kindred have undergone MAPT mutation analyses using both direct sequencing and BsrDI restriction enzyme digestion analyses (Fig. 6). The S305N mutation was identified in both affected sibs and absent in the unaffected members of the kindred. A C/A polymorphism at position 47 just before the start of exon 10 was found in both affected sisters and their father; by cloning this polymorphism was found to be on the normal allele in both affected sisters. This polymorphism is listed in the National Center for Biotechnology Information Single Nucleotide Polymorphism Database as rs3744460 and has an average heterozygosity of 0.467 (analysed in several Japanese samples). In addition, nine polymorphic markers (D3S1358, tetranucleotide repeats in the von Willebrand gene and the fibrinogen A a polypeptide gene, D8S1179, D21S11, D18S51, D5S818, D13S317, and D7S820) used in a standard paternity test were run in all five samples (Table 5). Results of these tests ruled against non-paternity. Over 250 clones were analysed from the mother and over 80 from the father and none were found to have the mutation, suggesting germline mosaicism. diagnosis was obvious when she began exhibiting behaviours similar to the proband. As shown in Tables 1 and 3, the sister was diagnosed at a much earlier stage of the illness, as reflected by the lower scores on the CDR, GDS and ROIL. Scores on the UPDRS have indicated parkinsonism in both siblings. Parkinsonism was far greater in the proband while she was receiving risperidone, and scores decreased upon its discontinuation. Neither sibling has developed features strongly suggestive of evolving corticobasal syndrome or progressive supranuclear palsy (Boeve et al., 2003), nor have features of motor neuron disease evolved. Analysis of neuropsychological data In both siblings, mental status test scores were within normal limits early in their course, and visuospatial/visuoconstructive skills were spared until late in the course. The proband performed poorly on the WCST early in her course, yet performance on several tests of attention/executive functioning has been minimally impaired or normal in the sister. Retention of (memory for) verbal information functioning and particularly confrontation naming were impaired in both siblings. Analysis of clinical data Analysis of neuropsychiatric inventory data The diagnosis of FTDP-17 in the proband was elusive until the neuropsychological and radiological studies suggested changes referable to the frontotemporal lobes. Her sister’s In the proband (Table 2), the NPI Total (sum of frequency 3 severity) ranged from 24 to 56 over this interval (30–45–56– 32–52–24–34–25). This is in spite of an increasing burden of 764 B. F. Boeve et al. A B C D Fig. 4 Coronal T1-weighted MRIs of case II.1 (sister) at ages 37 years 3 months (A), 37 years 9 months (B), 38 years 3 months (C) and 38 years 9 months (D). Atrophy is initially present in the amygdalae. Over time the mesial and inferior anterior temporal structures and mesial dorsolateral prefrontal and orbitofrontal cortex atrophy further, and the lateral ventricles enlarge (particularly the temporal horns). FTDP-17 associated with the S305N tau mutation 765 A B C D Fig. 5 Axial FLAIR MRIs of case II.1 (sister) at ages 37 years 3 months (A), 37 years 9 months (B), 38 years 3 months (C) and 38 years 9 months (D). The distribution of atrophy and lack of signal changes are quite similar to the proband’s FLAIR MRI studies (Fig. 3). 766 B. F. Boeve et al. A 400 300 200 100 383 252 131 1 2 3 4 5 6 Fig. 6 BsrDI restriction enzyme digestion of amplified MAPT exon 10 products from family members. Amplified products were run on a 2% agarose gel. Lane 1 contains 100 bp ladder DNA marker (Seegene, Seoul, Korea). The two affected sisters (lanes 2 and 4) have bands at 252 and 131 bp signifying the presence of the S305N allele. B Table 5 Alleles of polymorphic markers analysed in family members Locus Father Mother D31358 vWA FGA D8S1179 D21S11 D18S51 D5S818 D13S317 D7S820 16 15, 19 23 8, 13 31.2, 32.2 16, 17 11 11, 12 8, 12 17, 17 22, 12, 30, 12, 11 12, 10 18 Proband* Sibling 1* Sibling 2 16, 17, 25 22, 13 12, 32.2 30, 16 12, 11 13 11, 10, 17 19 23 13 31.2 16 13 12 16, 18 17, 19 22, 23 8, 12 31.2, 32.2 12, 16 11 12 8, 10 16, 18 17, 19 23, 25 13 30, 31.2 16, 17 11 11, 13 8, 10 *Individuals with frontotemporal dementia. psychotropic medications intended to address the behavioural disturbances. Apathy/indifference was present from the outset, and this has remained the most pervasive feature. Emotional lability is also present, with euphoria being present at one point, anxiety varying throughout her course, in spite of continued apathy. Sleep/wake problems were problematic early in her course, and some degree of aberrant motor behaviour and agitation has been present throughout. Hyperphagia was present at the initial NPI assessment. Disinhibition was perceived to be mild and minimally distressing. The emotional distress perceived by the proband’s husband over this interval (10–20–21–12–10–4–5–0) was maximal early in the course, and decreased over the most recent 212 years. The drop in emotional distress despite continued neuropsychiatric symptomatology suggests that the husband has adapted, developed tolerance and become a savvy caregiver. These may all be the same process. In the proband’s sister (Table 4), the mother’s ratings on the NPI Total ranged from 8 to 31 over this interval (10–8–31– 28), and the emotional distress varied in a similar fashion (11–3–13–6). The sister has exhibited euphoria, disinhibition Fig. 7 Graphs depicting longitudinal whole-brain volume (WBV) in ml (A) and ventricular volume (VV) in ml (B) over time for the proband and sister. Note that the plots are quite linear and the initial WBV values are similar for the two cases, but the initial VV values and slopes are quite different. and irritability from the outset, with depression, apathy and hyperphagia developing as the illness progressed. At no time during their periods of assessment were delusions or hallucinations present in either sibling. Analysis of radiological data Both siblings have developed striking atrophy of the anterior part of temporal lobes and moderate atrophy of the dorsolateral frontal, and orbitofrontal cortices, which in both cases is relatively symmetrical (Figs 2–5). As shown in Fig. 7, the plots of WBV (A) and VV (B) are quite linear, and the initial WBV values are similar. Yet the initial VV values and the slopes for WBV and VV are different. The annualized changes in WBV and VV, respectively, were –35.2 ml/year (3.23% FTDP-17 associated with the S305N tau mutation decrease per year) and +20.75 ml/year (16.93% increase per year) for the proband, and 30.75 ml/year (2.77% decrease per year) and +5.01 ml/year (3.11% increase per year) for the sister. Discussion Clinical and neuropsychiatric issues We present two young Caucasian siblings of European descent with many clinical, neuropsychological and radiological features typical of FTDP-17 (onset ages 30 and 36 years). While cognition is clearly impaired in both siblings, the most prominent aspect of their illness has been the neuropsychiatric features which heralded the onset and have proven challenging to manage despite aggressive pharmacological manipulations. Mild parkinsonism is also present in the proband, but this is probably due partly to risperidone as the parkinsonism has improved upon its discontinuation; subtle parkinsonism also exists in her sister. No features of the corticobasal syndrome (Boeve et al., 2003) or motor neuron disease have developed in these siblings and, interestingly, frontal release signs faded as the illness progressed in both cases. While the initial features in the proband were typical of FTDP-17, a familial disorder was not suggested by her family history at the time of her presentation. This case exemplifies that a genetic disorder may be at play even without a compatible family history. The CSF findings in the proband are also notable, as the results indicated that patients with a tauopathy can have a CSF tau and Ab profile consistent with Alzheimer’s disease (Galasko et al., 1998). There are conflicting data on CSF tau and Ab levels in FTD, some investigators finding elevations in tau compared with controls (Arai et al., 1997; Green et al., 1999; Molina et al., 1999; Fabre et al., 2001; Vanmechelen et al., 2001; Riemenschneider et al., 2002; Rosso et al., 2003) and others not (Mecocci et al., 1998; Sjögren et al., 2000a, b, 2001). CSF tau has been consistently elevated in Alzheimer’s disease, often exceeding levels in FTD (Arai et al., 1997; Green et al., 1999; Molina et al., 1999; Sjögren et al., 2000a, b; Vanmechelen et al., 2001; Riemenschneider et al., 2002). CSF Ab levels have tended to be lower in Alzheimer’s disease compared with FTD (Sjögren et al., 2000a; Vanmechelen et al., 2001; Riemenschneider et al., 2002). Even among patients with FTD associated with tau mutations, total tau and phosphorylated tau T181 levels were normal or only mildly increased (Rosso et al., 2003). These discrepancies in CSF tau levels in FTD compared with controls and Alzheimer’s disease may reflect methodological (e.g. technique for quantifying tau) or sampling (e.g. percentages of FTD patients with tauopathies) differences. The three affected relatives in the report of Iijima and colleagues had ages of onset ranging from 29 to 38 years, with personality changes evident early in the illness followed by cognitive decline, and parkinsonism was described as 767 minimal in one case and absent in the others (Iijima et al., 1999). Neuroimaging studies indicated left greater than right frontotemporal atrophy. There is no mention of symptoms or signs of motor neuron disease in this report, nor discussion of any of the typical clinical findings of the CBS despite the pathological findings in the proband being most consistent with corticobasal degeneration (CBD). We have specifically sought to identify signs typical of CBS and all have been absent thus far, which is not surprising as clinicopathological heterogeneity is known to exist in corticobasal degeneration (Boeve et al., 1999). In the only other reported kindred with the S305N tau mutation, the proband and his brother exhibited cognitive and behavioural changes typical of FTDP-17 (Kobayashi et al., 1999, 2002, 2003, 2004). The proband began exhibiting aggressive tendencies and personality changes at age 34, followed by disinhibited and stereotyped behaviours. Computed tomography of the head showed frontal lobe atrophy. He subsequently developed abulia, echolalia and limb rigidity that was levodopa-unresponsive. Terminally he displayed quadriplegia in flexion with rigidity, spasticity, and frontal release signs present prior to his death at age 46. Neuropathological examination revealed greater temporal than frontal lobe atrophy, profound nigral degeneration, and numerous Pick bodies and Pick cells compatible with the diagnosis of Pick’s disease (Kobayashi et al., 1999). This patient’s younger brother subsequently exhibited similar behavioural and cognitive changes beginning at age 39, and had MRI evidence of asymmetrical temporal–occipital cortical and hippocampal atrophy, with minimal frontal lobe atrophy (Kobayashi et al., 2002). Neuropathologically, taupositive Pick-like inclusions were identified (Kobayashi et al., 2003, 2004). Thus our cases appear clinically similar to the few described with this mutation, but the radiological features are slightly different, with only slight asymmetrical temporal > frontal atrophy in our two patients but asymmetrical temporal > frontal atrophy in the Japanese cases (Iijima et al., 1999; Kobayashi et al., 1999, 2002, 2003, 2004). Neuropsychological issues Based on previous studies, the expected neuropsychological features in the frontal variant or behavioural/dysexecutive subtype of FTD would include impaired performance on tests sensitive to frontal lobe functions (e.g. Trail-Making Test, Stroop, COWAT, WCST) and minimal or no impairment on tests assessing anterograde memory functioning (e.g. Wechsler Memory Scale—Third Edition, Wechsler Memory Scale—Logical Memory, Wechsler Memory Scale—Visual Reproductions, Free and Cued Selective Reminding Test) and visuospatial functioning (e.g. clock drawing, Rey–Osterrieth complex figure, Judgement of Line Orientation, Visual Form Discrimination) (Miller et al., 1991; Pachana et al., 1996; Neary et al., 1998; Hodges, 2001; Rascovsky et al., 2002). Performance on screening (e.g. MMSE, STMS) and global (e.g. DRS) tests of cognitive functioning are often normal or near normal early in the course (Gregory et al., 1997, 1999; 768 B. F. Boeve et al. Rahman et al., 1999). At the initial assessments, our cases indeed performed quite well on screening and global tests of cognition as well as on most tests of visuospatial functioning (Tables 1 and 3). In fact over the 2 years of assessments on the sister, she has scored in the normal range on the MMSE and STMS, and her total score on the DRS has achieved marginal impairment on only one round of testing (at least when compared with normative data on individuals many years older than she). The proband was unable to achieve all six categories on the WCST and had numerous perseverative errors, typical of FTD (the WCST is not part of our research protocol and therefore the sister has not been assessed on this measure). The sister has scored in the impaired range on the Digit Span, Digit Symbol-coding, and Similarities subtests of the WAISIII, as well as attention subtest of the DRS, while performance on other tests of attention/executive functioning has been minimally impaired or normal. While the neuropsychiatric features have dominated the sister’s clinical course, with only mild forgetfulness occurring in everyday activities, her performance on some tests of anterograde memory functioning has been poor. Although reports of severe memory impairment in FTD do exist (Caine et al., 2001), performance on delayed recall measures is typically minimally affected early in the course of FTD (Gregory et al., 1997, 1999; Rahman et al., 1999; Pasquier et al., 2001). Although considerable data have been accumulated on memory measures in FTD, no score on any memory measure has been accepted as necessary to warrant or negate the diagnosis of FTD. Poor performance on memory measures can also reflect poor attention, or impaired semantic processing on verbal memory measures. The proband and particularly the sister therefore exemplify how some patients with FTD associated with a presumed pathogenic mutation in tau can perform poorly on episodic memory measures. We suspect the hippocampal atrophy now present in both siblings is the radiological correlate for their memory impairment, which is consistent with another reported FTD patient (Pickering-Brown et al., 2004). We cannot exclude some degree of medication effect contributing to impairment on some measures in both siblings. Receptive aphasia and agnosia have not been prominent problems in either sibling’s clinical course, yet both have performed poorly on confrontation naming tests (e.g. MLAE naming, Boston Naming Test) even at their initial assessments. Since integrity of the anterior inferolateral temporal region in the dominant hemisphere is necessary for intact confrontation naming, and this region is typically affected early in the course in frontal variant FTD as well as temporal variant FTD (Edwards-Lee et al., 1997; Hodges and Miller, 2001b), poor performance in confrontation naming in the setting of more normal performance on other measures (particularly memory) may suggest underlying FTD rather than AD (Boone et al., 1999; Hodges et al., 1999; Miller and Gearhart, 1999; Hodges and Miller, 2001b; Boccardi et al., 2003). Progressive language dysfunction, which certainly occurs to some degree in most FTD patients, can lead to what may appear to be a rapid rate of progression based on clinical and neuropsychological assessments. Since performance on most clinical and neuropsychological measures is dependent on relatively preserved language functioning, an aphasic disturbance can lead to poor performance on measures assessing non-language domains. The proband’s progressive aphasia probably contributed to her notable decline on clinical and neuropsychological measures. Radiological issues As noted above, our patients’ MRI findings are similar to those of the few other S305N tau mutation cases, with temporal greater than frontal lobe atrophy, although atrophy was largely symmetrical in our cases and markedly asymmetrical in the other cases (Iijima et al., 1999; Kobayashi et al., 1999, 2002, 2003, 2004). The MR findings in our siblings are also quite typical of FTD (Miller and Gearhart, 1999; Hodges, 2001; Boccardi et al., 2002, 2003; Rosen et al., 2002). Most reports note asymmetrical atrophy in FTD patients, although one report specifically investigated symmetrical frontotemporal atrophy in sporadic FTD cases (Boccardi et al., 2002). Compared with FTD patients with asymmetrical brain atrophy, those with symmetrical atrophy had an earlier age of onset, more severe global and medial temporal atrophy, and a greater frequency of ApoE e4 alleles, yet this report involves 10 FTD patients of whom three possessed an ApoE e4 allele (Boccardi et al., 2002). Our siblings indeed had an early age of onset, but neither had ApoE e4 alleles. A much larger number of FTD cases with symmetrical and asymmetrical atrophy, with and without ApoE e4 alleles, will be necessary to determine if any association exists between brain atrophy in FTD and the presence or absence of the ApoE e4 allele. The longitudinal volumetric MRI findings in our two patients permit some interesting observations. The slightly greater WBV in the sister compared with the proband is consistent with the clinical and neuropsychological data reflecting less impairment in the sister at the time of her first scan. Yet the VV is much greater in the sister than the proband, which is not readily explainable. The rate of decline in WBV and the rate of increase in VV are far greater in the proband than in the sister, which is also consistent with the rather minimal clinical and neuropsychological changes over time in the sister compared with the more dramatic changes in the proband. Yet why these rates are so different is not clear. Both patients presumably have the same disease, both have many of the same clinical and neuropsychiatric features, and both have been treated with almost identical treatment regimens. Whether drug intervention earlier in the illness in the sister afforded her a slower rate of progression or whether some other environmental or biological factors are at play cannot be determined at this time. However, variability in the phenotypes, ages of onset and durations of disease is well known among relatives with the same genetic mutation in familial neurodegenerative disorders. This issue has implications for FTDP-17 associated with the S305N tau mutation treatment trials (see below). Furthermore, variable penetrance or non-penetrance could affect the phenotypic expression; non-penetrance has been reported in tau mutations (Murrell et al., 1999; Pickering-Brown et al., 2002; van Herpen et al., 2003). We and others have been refining techniques to quantify rates of regional or whole-brain atrophy using MRI in patients with mild cognitive impairment and Alzheimer’s disease (Jack et al., 1998, 2000, 2003; Fox et al., 1999, 2000; Gunter et al., 2003), and more recently in patients with FTD (Chan et al., 2001a, b). The automated methods, such as the boundary shift integral method used in our patients and the gradient-matching method, allow excellent precision and high throughput (Gunter et al., 2003), and such methods look promising as surrogate markers of disease progression for naturalistic (such as the findings reported herein) and therapeutic trials. The fact that the relationship is linear in most patients bodes well for developing and interpreting therapeutic drug trials. Genetic issues Since its initial description, several investigators have determined that the exonic S305N mutation is unique in that it does not result in the impairment of the tau–microtubule binding but in the overproduction of four-repeat isoforms through increased splicing-in of exon 10 and destabilization of the RNA stem–loop structure, an abnormality characteristic of intronic mutations in the MAPT gene (Grover et al., 1999; Hasegawa et al., 1999; Varani et al., 1999). Another mutation in exon 10 (N279K) acts in a similar fashion (D’Souza et al., 1999; Hasegawa et al., 1999). More recently, a S305S tau mutation associated with progressive supranuclear palsy clinical and pathological features has been reported (Stanford et al., 2000). Although this mutation does not result in an amino acid change, splicing of exon 10 is significantly increased (Stanford et al., 2000). Thus, the S305N mutation in our cases is very likely pathogenic. The polymorphism on the normal MAPT allele (position 47, adenine for cytosine) in the affected sibs and their father suggests that he is the biological father. The additional analyses (Table 5) confirm that he is indeed the biological father, thus arguing against non-paternity in this family. Incomplete penetrance cannot explain the findings in this kindred as neither parent has the S305N mutation. Since the likelihood of an identical spontaneous mutation occurring in two siblings is extremely low, we suspect germline mosaicism is the most likely explanation for the findings in this kindred. Somatic mosaicism was recently reported in a subject with earlyonset Alzheimer’s disease (Beck et al., 2004). The subject showed signs of disease in the mid 50s and had a daughter who showed signs at age 27. The daughter was found to have a mutation in the Presenilin 1 gene (P436Q). After further testing, it was shown that the mother did have the mutation in a few of her somatic cells. Gonadal mosaicism was reported in one unaffected mother who had two daughters with repeat 769 expansions of the CAG trinucleotide repeat in the Huntington disease gene (Laccone and Christian, 2000). Germline mosaicism has never been reported in the parents of an individual with a MAPT mutation to our knowledge. Implications for future drug trials The longitudinal findings in these siblings are particularly relevant for future experimental drug trials in FTD and the tauopathies. Much of the research on FTD has centred on features that discriminate FTD from other dementias (Gregory and Hodges, 1996; Mendez et al., 1996, 1998; Pachana et al., 1996; Gregory et al., 1997; Lindau et al., 2000; Mummery et al., 2000; Perry and Hodges, 2000; Hodges and Miller, 2001a; Lough et al., 2001; Boxer et al., 2003; Nyatsanza et al., 2003). While this work is certainly important for improving the diagnosis of FTD, there is little published data on the longitudinal course of FTD (Gregory, 1999; Chan et al., 2001a), nor is there much data that help guide the future treatment of patients with FTD. To date there have been few reports on the symptomatic management of neuropsychiatric manifestations of FTD (Swartz et al., 1997), and since no agent has yet been identified that significantly impacts tau protein pathophysiology, no treatment trial for FTDP-17 or the sporadic tauopathies has begun. Developing treatment trials will be challenging for several reasons. The disorders that can manifest as the syndrome of FTD include Pick’s disease, corticobasal degeneration, progressive supranuclear palsy, frontotemporal lobar degeneration with and without motor neuron disease-like inclusions, neurofilament inclusion body disease, and rarely Creutzfeldt–Jakob disease and Alzheimer’s disease (Hodges and Miller, 2001a; McKhann et al., 2001; Rossor, 2001; Boeve et al., 2003; Josephs et al., 2003). Hence, treatments active against tau pathophysiology may be effective for one or more of the tauopathies, but may not be effective for frontotemporal lobar degeneration with or without MND since most cases do not have tau-positive pathology. Furthermore, even among the tauopathies drug development will be challenging to assess, as there are cognitive, neuropsychiatric and motor manifestations that must be monitored, and relatives with the same mutation may exhibit quite different features. When considering how efficacy will be determined in any treatment trial, one would want to make measurements of various parameters over time and determine if the intervention will delay the rate of progression or halt progression altogether (Fig. 8). The parameters to be assessed could include clinical (e.g. MMSE, UPDRS), functional (e.g. GDS, ROIL), neuropsychological (e.g. DRS, TMT), neuropsychiatric (e.g. NPI scales) and radiological (e.g. WBV, VV, hippocampal volume) measures. One would need to assess a sufficient number of patients with FTD with a standardized battery of tests over time to determine which parameters change in the most consistent and linear fashion. In our two cases, the parameters that appeared to trend in the most consistent manner were the ROIL, WBV and VV. Using the theoretical construct shown 770 B. F. Boeve et al. normal d c measure b a abnormal time Fig. 8 Graph depicting theoretical constructs for considering a treatment trial, where the values of any measure ( y axis) are plotted against time (x axis). The arrow represents the onset of treatment, line ‘a’ represents the course in the untreated setting, line ‘b’ represents the course if the treatment decreases the rate of progression, line ‘c’ represents the course if the treatment halts progression altogether, and line ‘d’ represents the course if treatment results in improvement (e.g. neuropsychological measure, NPI measure). in Fig. 8, patients with FTD features could act as their own controls, with the hope that therapy would alter the rate of change (as in line b or c in Fig. 8). Because members of a kindred with the same mutation can exhibit different features and rates of change (as demonstrated by our two siblings), and the same is true of sporadic FTD and some tauopathies, one important question is whether to design clinical trials with placebo and active drug arms and compare rates of change by groups (placebo versus active drug), or use crossover methodology with patients using placebo for one period of assessment and active drug for another period of assessment, and then comparing rates of change within each patient. Clearly more patients will need to be assessed in a standardized longitudinal manner to optimally develop treatment trial methodologies appropriate for patients with FTDP-17 and the sporadic tauopathies. We are hopeful that data such as we report herein may aid investigators to develop methodology; many of the cognitive, neuropsychological, neuropsychiatric, motor and radiological findings discussed in this report are being assessed in longitudinal FTD study funded by the National Institute on Aging (RO1 AG23195). Acknowledgements This study was supported by grants P50 AG 16574 (Mayo Alzheimer’s Disease Research Center) P30 AG10133 (Indiana Alzheimer Disease Center) and R01 NS 37431 (J. M.). We thank the staff of the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation, Indiana Alzheimer’s Disease Research Center, University of Wisconsin Departments of Neurology and Genetics, and Monroe Clinic for assisting with the collection of data and care of the patients and families. We also thank Dr David Knopman for his critical review of this paper. We are particularly grateful to the patients and their families for participating in this research. References Arai H, Morikawa Y, Higuchi M, Matsui T, Clark CM, Miura M, et al. Cerebrospinal fluid tau levels in neurodegenerative diseases with distinct tau-related pathology. Biochem Biophys Res Commun 1997; 236: 262–4. Beck JA, Poulter M, Campbell T, Uphill JB, Adamson G, Geddes JF, et al. Nuclear somatic mosaicism in sporadic early-onset alzheimer’s disease. Neurobiol Aging 2004; 25 (S2): S54. Benton A, Hamsher KD. Multilingual Aphasia Examination (Manual). Iowa City (IA): University of Iowa; 1978. Benton A, Hamsher KD, Varney N, Spreen O. Contributions to neuropsychological assessment. New York (NY): Oxford University Press; 1983. Bird T, Knopman D, VanSwieten J, Rosso S, Feldman H, Tanabe H, et al. Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol 2003; 54 (Suppl. 5): S29–31. Boccardi M, Laakso MP, Bresciani L, Geroldi C, Beltramello A, Frisoni GB. Clinical characteristics of frontotemporal patients with symmetric brain atrophy. Eur Arch Psychiatr Clin Neurosci 2002; 252: 235–9. Boccardi M, Laakso MP, Bresciani L, Galluzzi S, Geroldi C, Beltramello A, et al. The MRI pattern of frontal and temporal brain atrophy in frontotemporal dementia. Neurobiol Aging 2003; 24: 95–103. Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999; 53: 795–800. Boeve B, Lang A, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54: S15–19. Boone KB, Miller BL, Lee A, Berman N, Sherman D, Stuss DT. Neuropsychological patterns in right versus left frontotemporal dementia. J Int Neuropsychol Soc 1999; 5: 616–22. Boxer AL, Rankin KP, Miller BL, Schuff N, Weiner M, Gorno-Tempini ML, et al. Cinguloparietal atrophy distinguishes Alzheimer disease from semantic dementia. Arch Neurol 2003; 60: 949–56. Buschke H. Cued recall in amnesia. J Clin Neuropsychol 1984; 6: 433–40. Caine D, Patterson K, Hodges JR, Heard R, Halliday G. Severe anterograde amnesia with extensive hippocampal degeneration in a case of rapidly progressive frontotemporal dementia. Neurocase 2001; 7: 57–64. Chan D, Fox NC, Jenkins R, Scahill RI, Crum WR, Rossor MN. Rates of global and regional cerebral atrophy in AD and frontotemporal dementia. Neurology 2001a; 57: 1756–63. Chan D, Fox NC, Scahill RI, Crum WR, Whitwell JL, Leschziner G, et al. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer’s disease. Ann Neurol 2001b; 49: 433–42. Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994; 44: 2308–14. D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee V, Bird T, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism– chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 1999; 96: 5598–603. Edwards-Lee T, Miller BL, Benson DF, Cummings JL, Russell GL, Boone K, et al. The temporal variant of frontotemporal dementia. Brain 1997; 120: 1027–40. Fabre SF, Forsell C, Viitanen M, Sjogren M, Wallin A, Blennow K, et al. Clinic-based cases with frontotemporal dementia show increased cerebrospinal fluid tau and high apolipoprotein E epsilon4 frequency, but no tau gene mutations. Exp Neurol 2001; 168: 413–8. Fahn S, Elton R, Committee Mot UD. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden C, Calne D and Goldstein M, editors. Recent developments in Parkinson’s disease, Vol. 11. Florham Park (NJ): MacMillan; 1987. p. 153–63. FTDP-17 associated with the S305N tau mutation Folstein M, Folstein S, McHugh P. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98. Fox NC, Warrington EK, Rossor MN. Serial magnetic resonance imaging of cerebral atrophy in preclinical Alzheimer’s disease. Lancet 1999; 353: 2125. Fox NC, Cousens S, Scahill R, Harvey RJ, Rossor MN. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol 2000; 57: 339–44. Galasko D, Chang L, Motter R, Clark C, Kaye J, Knopman D, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol 1998; 55: 937–45. Ghetti B, Hutton M, Wszolek ZK. Frontotemporal dementia and parkinsonism linked to chromosome 17 associated with Tau gene mutations (FTDP-17T). In: D. Dickson (Ed) Neurodegeneration. The Molecular Pathology of Dementia and Movement Disorders. 2003; pp 86–102. Green A, Harvey R, Thompson E, Rossor M. Increased tau in the cerebrospinal fluid of patients with frontotemporal dementia and Alzheimer’s disease. Neurosci Lett 1999; 259: 133–5. Gregory C. Frontal variant of frontotemporal dementia: a cross-sectional and longitudinal study of neuropsychiatric features. Psychol Med 1999; 29: 1205–7. Gregory CA, Hodges JR. Clinical features of frontal lobe dementia in comparison to Alzheimer’s disease. J Neural Transm 1996; 47: 103–23. Gregory CA, Orrell M, Sahakian B, Hodges JR. Can frontotemporal dementia and Alzheimer’s disease be differentiated using a brief battery of tests? Int J Geriatr Psychiatry 1997; 12: 375–83. Gregory CA, Serra-Mestres J, Hodges JR. Early diagnosis of the frontal variant of frontotemporal dementia: how sensitive are standard neuroimaging and neuropsychologic tests? Neuropsychiatr Neuropsychol Behav Neurol 1999; 12: 128–35. Grober E, Buschke H. Genuine memory deficits in dementia. Dev Neuropsychol 1987; 3: 13–36. Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, et al. 50 splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem 1999; 274: 15134–43. Gunter J, Shiung M, Manduca A, Jack CJ. Methodological considerations for measuring rates of brain atrophy. J Magn Reson Imaging 2003; 18: 16–24. Hasegawa M, Smith M, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett 1999; 443: 93–6. Heaton R. Wisconsin Card Sorting Test Manual. Odessa (FL): Psychological Assessment Resources; 1981. Hodges JR. Frontotemporal dementia (Pick’s disease): clinical features and assessment. Neurology 2001; 56: S6–10. Hodges JR, Miller B. The classification, genetics and neuropathology of frontotemporal dementia. Introduction to the special topic papers: part I. Neurocase 2001a; 7: 31–5. Hodges JR, Miller B. The neuropsychology of frontal variant frontotemporal dementia and semantic dementia. Introduction to the special topic papers: part II. Neurocase 2001b; 7: 113–21. Hodges JR, Patterson K, Ward R, Garrard P, Bak T, Perry R, et al. The differentiation of semantic dementia and frontal lobe dementia (temporal and frontal variants of frontotemporal dementia) from early Alzheimer’s disease: a comparative neuropsychological study. Neuropsychology 1999; 13: 31–40. Iijima M, Tabira T, Poorkaj P, Schellendberg G, Trojanowski J, Lee V, et al. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport 1999; 10: 497–501. Jack CR Jr, Petersen RC, Xu Y, O’Brien PC, Smith GE, Ivnik RJ, et al. Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology 1998; 51: 993–9. 771 Jack CR Jr, Petersen RC, Xu Y, O’Brien PC, Smith GE, Ivnik RJ, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 2000; 55: 484–9. Jack CR Jr, Slomkowski M, Gracon S, Hoover TM, Felmlee JP, Stewart K, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology 2003; 60: 253–60. Josephs K, Holton J, Rossor M, Braendgaard H, Ozawa T, Fox N, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain 2003; 126: 2291–303. Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Boston (MA): E. Kaplan & H. Goodglass; 1978. Kaufman A, Kaufman N. Kaufman Brief Intelligence Test Manual. Circle Pines (MN): American Guidance Service; 1990. Kobayashi K, Hayashi M, Fukutani Y, Miyazu K, Shiozawa M, Muramori F, et al. KP1 expression of ghost Pick bodies, amyloid P-positive astrocytes and selective nigral degeneration in early onset Pick’s disease. Clin Neuropathol 1999; 18: 240–50. Kobayashi K, Hayashi M, Kidani T, Nakano H, Miyazu K, Ujike H, et al. Pick’s disease in 2 brothers with S305N mutation: note in supplement to an earlier communication. Clin Neuropathol 2002; 21: 191–3. Kobayashi K, Kidani T, Ujike H, Hayashi M, Ishihara T, Miyazu K, et al. Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17) with a missense mutation of S305N closely resembling Pick’s disease. J Neurol 2003; 250: 990–2. Kobayashi K, Hayashi M, Kidani T, Ujike H, Iijima M, Ishihara T, et al. Pick’s disease pathology of a missense mutation of S305N of frontotemporal dementia and parkinsonism linked to chromosome 17: another phenotype of S305N. Dement Geriatr Cogn Disord 2004; 17: 293–7. Kokmen E, Smith G, Petersen R, Tangalos E, Ivnik R. The short test of mental status: correlations with standardized psychometric testing. Arch Neurol 1991; 48: 725–8. Laccone F, Christian W. A recurrent expansion of a maternal allele with 36 CAG repeats causes Huntington disease in two sisters. Am J Hum Genet 2000; 66: 1145–8. Lindau M, Almkvist O, Kushi J, Boone K, Johansson SE, Wahlund LO, et al. First symptoms—frontotemporal dementia versus Alzheimer’s disease. Dement Geriatr Cogn Disord 2000; 11: 286–93. Lough S, Gregory C, Hodges JR. Dissociation of social cognition and executive function in frontal variant frontotemporal dementia. Neurocase 2001; 7: 123–30. Mattis S. Dementia Rating Scale: Professional Manual. Odessa (FL): Psychological Assessment Resources; 1988. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 2001; 58: 1803–9. Mecocci P, Cherubini A, Bregnocchi M, Chionne F, Cecchetti R, Lowenthal D, et al. Tau protein in cerebrospinal fluid: a new diagnostic and prognostic marker in Alzheimer disease? Alzheimer Dis Assoc Disord 1998; 12: 211–4. Members of the Department of Neurology at the Mayo Clinic. Clinical Examinations in Neurology. St Louis (MO): Mosby–Year Book; 1998. Mendez MF, Cherrier M, Perryman KM, Pachana N, Miller BL, Cummings JL. Frontotemporal dementia versus Alzheimer’s disease: differential cognitive features. Neurology 1996; 47: 1189–94. Mendez MF, Perryman KM, Miller BL, Cummings JL. Behavioral differences between frontotemporal dementia and Alzheimer’s disease: a comparison on the BEHAVE-AD rating scale. Int Psychogeriatr 1998; 10: 155–62. Miller BL, Gearhart R. Neuroimaging in the diagnosis of frontotemporal dementia. Dement Geriatr Cogn Disord 1999; 10: 71–4. Miller BL, Cummings JL, Villanueva-Meyer J, Boone K, Mehringer CM, Lesser IM, et al. Frontal lobe degeneration: clinical, neuropsychological, and SPECT characteristics. Neurology 1991; 41: 1374–82. 772 B. F. Boeve et al. Miller BL, Diehl J, Freedman M, Kertesz A, Mendez M, Rascovsky K. International approaches to frontotemporal dementia diagnosis: from social cognition to neuropsychology. Ann Neurol 2003; 54: S7–10. Molina L, Touchan J, Herpe M, Lefranc D, Duplan L, Cristol J, et al. Tau and apo E in CSF: potential aid for discriminating Alzheimer’s disease from other dementias. Neuroreport 1999; 10: 3491–5. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43: 2412–4. Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges JR. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol 2000; 47: 36–45. Munoz DG, Dickson D, Bergeron C, Mackenzie IR, Delacourte A, Zhukareva V. The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol 2003; 54: S24–28. Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, et al. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol 1999; 58: 1207–26. Neary D, Snowden J, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546–54. Northern California Neurobehavioral Group. Neurobehavioral Cognitive Status Examination. Fairfax (CA): Northern California Neurobehavioral Group, Inc.; 1988. Nyatsanza S, Shetty T, Gregory C, Lough S, Dawson K, Hodges JR. A study of stereotypic behaviours in Alzheimer’s disease and frontal and temporal variant frontotemporal dementia. J Neurol Neurosurg Psychiatr 2003; 74: 1398–402. Osterrieth P. Le test de copie d’une figure complexe. Arch Psychol 1944; 30: 206–356. Pachana NA, Boone KB, Miller BL, Cummings JL, Berman N. Comparison of neuropsychological functioning in Alzheimer’s disease and frontotemporal dementia. J Int Neuropsychol Soc 1996; 2: 505–10. Pasquier F, Grymonprez L, Lebert F, Van der Linden M. Memory impairment differs in frontotemporal dementia and Alzheimer’s disease. Neurocase 2001; 7: 161–71. Perry RJ, Hodges JR. Differentiating frontal and temporal variant frontotemporal dementia from Alzheimer’s disease. Neurology 2000; 54: 2277–84. Pickering-Brown S, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain 2004; 127: 1415–26. Pickering-Brown SM, Richardson AMT, Snowden JS, M McDonagh A, Burns A, Braude W, et al. Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 2002; 125: 732–51. Rahman S, Sahakian BJ, Hodges JR, Rogers RD, Robbins TW. Specific cognitive deficits in mild frontal variant frontotemporal dementia. Brain 1999; 122: 1469–93. Rascovsky K, Salmon DP, Ho GJ, Galasko D, Peavy GM, Hansen LA, et al. Cognitive profiles differ in autopsy-confirmed frontotemporal dementia and AD. Neurology 2002; 58: 1801–8. Reed L, Wszolek Z, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging 2001; 22: 89–107. Reisberg B, Ferris S, deLeon M, Crook T. Global deterioration scale (GDS). Psychopharmacol Bull 1988; 24: 661–2. Reitan R. Validity of the Trail-Making Test as an indication of organic brain damage. Percept Mot Skills 1958; 8: 271–6. Rey A. L’examen psychologique dan les cas d’encephalopathie traumatique. Arch Psychol 1941; 28: 286–40. Rey A. L’examen clinique en psychologie. Paris: Presses Universitaires de France; 1964. Riemenschneider M, Wagenpfeil S, Diehl J, Lautenschlager N, Theml T, Heldmann B, et al. Tau and Abeta42 protein in CSF of patients with frontotemporal degeneration. Neurology 2002; 58: 1622–8. Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002; 58: 198–208. Rosso SM, van Herpen E, Pijnenburg YA, Schoonenboom NS, Scheltens P, Heutink P, et al. Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch Neurol 2003; 60: 1209–13. Rossor M. Pick’s disease: a clinical overview. Neurology 2001; 56 (Suppl. 4): S3–5. Sjögren M, Minthon L, Davidsson P, Granerus AK, Clarberg A, Vanderstichele H, et al. CSF levels of tau, beta-amyloid(1–42) and GAP-43 in frontotemporal dementia, other types of dementia and normal aging. J Neural Transm 2000a; 107: 563–79. Sjögren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology 2000b; 54: 1960–4. Sjögren M, Davidsson P, Gottfries J, Vanderstichele H, Edman A, Vanmechelen E, et al. The cerebrospinal fluid levels of tau, growthassociated protein-43 and soluble amyloid precursor protein correlate in Alzheimer’s disease, reflecting a common pathophysiological process. Dem Geriatr Cog Disord 2001; 12: 257–64. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 1998; 95: 7737–41. Stanford P, Halliday G, Brooks W, Kwok J, Storey C, Creasey H, et al. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 2000; 123: 880–93. Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatr 1997; 58: 212–6. Tang-Wai DF, Knopman DS, Geda YE, Edland SD, Smith GE, Ivnik RJ, et al. Comparison of the short test of mental status and the mini-mental state examination in mild cognitive impairment. Arch Neurol 2003; 60: 1777–81. Trenerry M, Crosson B, DeBoe J, Leber W. Stroop Neurological Screening Test (Manual). Odessa (FL): Psychological Assessment Resources; 1989. van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, van de Graaf R, et al. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 2003; 54: 573–81. Vanmechelen E, Vanderstichele H, Hulstaert F, Andreasen N, Minthon L, Winblad B, et al. Cerebrospinal fluid tau and beta-amyloid(1–42) in dementia disorders. Mech Ageing Dev 2001; 122: 2005–11. Varani L, Hasegawa M, Spillantini M, Smith M, Murrell J, Ghetti B, et al. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci USA 1999; 96: 8229–34. Wechsler D. Wechsler Adult Intelligence Scale—Third Edition. San Antonio (TX): Psychological Corporation; 1997a. Wechsler D. Wechsler Memory Scale—Third Edition. San Antonio (TX): Psychological Corporation; 1997b. Weintraub S. The record of independent living: an informant-completed measure of activities of daily living and behavior in elderly patients with cognitive impairment. Am J Alzheimer Care Rel Disord 1986; 7: 35–9. Wilkinson G. WRAT3 Administration Manual. Wilmington (DE): Wide Range; 1993.