Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Orphan drug wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Psychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Theralizumab wikipedia , lookup
Neuropharmacology wikipedia , lookup
Drug design wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Pharmacognosy wikipedia , lookup
Prescription costs wikipedia , lookup
Drug discovery wikipedia , lookup
Drug interaction wikipedia , lookup
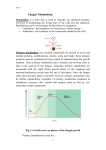
WelcometoWeek4 Startingweekfourvideo Please watch the online video (46 seconds). OPTIONAL‐Please participate in the online discussion forum. 7.7OralDeliveryII Multipleoraldosesvideo Please watch the online video (7 minutes, 34 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. CalculatingCpforanoraldrug Background: The Cp of an oral drug is equal to the sum of the remaining drug from all the previous doses administered to a patient. Instructions: Read the passage below on calculating Cp for a drug and use the information to answer the assessment questions that follow. Learning Goals: To understand better how to determine Cp values for oral drugs at different time points. The Cp for an oral drug dose can be calculated at any time with the formula below if all the variables are known. Any reference with pharmacokinetic parameters on drugs will list bioavailabilty (F), volume of distribution (Vd), half‐life (t1/2), and tmax, among others. kel can be calculated from t1/2. kab can be estimated from tmax and kel (with some trial and error). With these parameters in hand, one can calculate Cp at any time. In order to determine Cp from many doses, one must calculate the Cp from each dose and add them together. An important idea to remember is that the time of each dose is different. For example, if a drug is dosed every 4 hours, then at 10 hours, Cpdose 1 would be calculated at the full time of 10 hours, but Cpdose 2 would be calculated at only 6 hours, and Cpdose 3 would be calculated at just 2 hours. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. Complexitiesofdosing Background: A successful dosing regimen usually involves administering a drug so that the concentration of the drug remains within a window that provides a therapeutic effect without demonstrating toxic effects. Instructions: Read the case study on daptomycin, a drug that poses particularly difficult dosing challenges. Learning Goal: To gain exposure to more subtle issues surrounding the dosing of drugs. Daptomycin is a large antibiotic. Daptomycin violates Lipinski's rules in just about every way possible. Its molecular weight is 1,620 g/mol, well above the 500 g/mol limit of Lipinski. Daptomycin also has far more than 5 hydrogen bond donors and 10 hydrogen bond acceptors. Based on these traits, one might expect that daptomycin has a very low oral bioavailability. That expectation is correct. Daptomycin cannot be formulated as an oral drug and is instead administered intravenously. (See structure on following page.) The antibacterial activity of daptomycin was originally discovered by scientists at Eli Lilly in the 1980s. The therapeutic effect of daptomycin is accompanied with toxicity to muscle tissues. Unfortunately, the concentrations required for the antibacterial activity of daptomycin are very similar to those that cause muscle toxicity. Researchers at Eli Lilly explored several options for administering daptomycin and ultimately focused upon a twice daily IV regimen. The hope was that a twice daily dosing would provide a narrower range for Cp in the patient. By hitting a narrow Cp range, the drug may be able to hit concentrations ideal for antibacterial activity and yet avoid or minimize the toxic effects. A representation of Cp vs. time for both once daily (black line) and twice daily (red line) dosing is shown below. Note that the twice daily schedule does keep Cp of daptomycin within a tighter range. Unfortunately, researchers at Eli Lilly were unable to separate satisfactorily the antibacterial and toxic effects of daptomycin. Eli Lilly closed their daptomycin research program. Another company, Cubist, expressed interest in reviving the project. Eli Lilly then entered an agreement with Cubist, and Cubist started its own research program in daptomycin in 1997. This type of arrangement is common between drug companies. Large companies often have more potential projects than available resources. Projects that are not making progress will be shelved to free resources for more promising ones. Partnering with another company allows more projects to be in development. If the shared project is successful, then both companies will share in the profits. The drug discovery group at Cubist pursued a once daily dosing regimen for daptomycin. Instead of minimizing the peak Cp values of daptomycin with twice daily dosing, Cubist's approach emphasized the deeper Cp troughs of a once daily dosing. Cubist found that the muscle toxicity effects could be minimized without sacrificing antibacterial activity. One possible interpretation is that the deeper dips in Cp in the once daily regimen allow the muscles to recover periodically from the toxic effects of the drug. Cubist obtained a patent on their specific and novel dosing schedule which was counter to the prevailing logic on daptomycin. Even though the composition of matter patent on daptomycin has expired, generic manufacturers have been excluded from the market because of Cubist's dosing patent. The case of daptomycin demonstrates how each drug brings its unique challenges to drug discovery. The therapeutic and toxic levels of each drug create new issues that must be addressed by medicinal chemists. OPTIONAL‐Please participate in the online discussion forum. 7.8CLandVdRevisited CLvs.Vdplotsvideo Please watch the online video (7 minutes, 27 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. ModifyingstructurestoimprovePK Background: A CL vs. Vd plot is an excellent tool for providing insight into how a drug's structure might be modified in order to optimize its pharmacokinetics. Instructions: Answer the questions below. Learning Goal: To learn how to modify a drug or lead in order to achieve desired pharmacokinetic properties. Daptomycin is a large antibiotic. Daptomycin violates Lipinski's rules in just about every way possible. Its molecular weight is 1,620 g/mol, well above the 500 g/mol limit of Lipinski. Daptomycin also has far more than 5 hydrogen bond donors and 10 hydrogen bond acceptors. Based on these traits, one might expect that daptomycin has a very low oral bioavailability. That expectation is correct. Daptomycin cannot be formulated as an oral drug and is instead administered intravenously. (See structure on following page.) Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. Chapter8‐Metabolism IntroductiontoChapter8 Chapter 8 contains six subsections. Introduction Phase I Pt 1 Phase I Pt 2 Phase II Metabolite Issues Prodrugs Upon completing this chapter, you should understand the common types of drug metabolism reactions that occur in the body and how to recognize and predict those reactions. You should also recognize the complications that metabolites introduce to drug discovery based on demographic difference and biological activity of the metabolites. Finally, you will see drug metabolism as an exploitable process to improve the bioavailability of certain drugs. OPTIONAL‐Please participate in the online discussion forum. 8.1Introduction MetabolismandVdvideo Please watch the online video (7 minutes, 9 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Comparingmetabolites Background: When drugs are metabolized, the resulting metabolites are typically (but not always) more polar than the original drug. One simple method for comparing the polarity of two compounds is through lipophilicity, log P. In general, compounds with a lower lipophilicity (lower log P), have a lower Vd and a shorter half‐life. Instructions: Use data from the DrugBank (http://www.drugbank.ca/)to answer the questions below. Learning Goal: To understand more fully how metabolism can affect a drug and its metabolites in terms of polarity and Vd. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. 8.2PhaseIPtI Oxidationvideo Please watch the online video (6 minutes, 52 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Omegaoxidation Background: Most oxidations of sp3 hybridized carbons occur adjacent to a nitrogen or oxygen atom and cause dealkylation of the nitrogen or oxygen. Instructions: Read the passage below for an example of another type of sp3 hybridized carbon oxidation. Learning Goal: To see another somewhat common type of Phase I oxidation. Oxidations of simple sp3 hybridized carbon atoms are less rapid because the oxidation is not activated by an adjacent oxygen or nitrogen atom. Simple alkyl chains can regardless undergo metabolism in the body. These oxidations are called ω‐oxidations (omega oxidations). One classic example of an ω‐oxidation was seen in the previous subsection. Terfenadine, an antihistamine, undergoes ω‐oxidation on one of the tert‐butyl carbons. The C‐H bond is presumably oxidized to an alcohol, which is rapidly oxidized to an aldehyde and then the acid. Interestingly, the acid metabolite is itself also an antihistamine drug. Another example of an ω‐oxidation can be found in the antidiabetic chlorpropamide. The propyl chain in chlorpropamide actually undergoes two different ω‐oxidations. One is on the end of the propyl chain. This is a true ω‐oxidation as it occurs on the end (omega) of the chain. The other is an oxidation on the second‐to‐last carbon. This is formally called an ω‐1‐oxidation because it occurs one carbon removed from the end of the chain. While ω‐oxidations are less common than other types of Phase I metabolic reactions, they are encountered with some regularity. OPTIONAL‐Please participate in the online discussion forum. PredictingphaseImetabolites Background: Based on the functional groups in a molecule, the most likely metabolites of a drug can often be predicted. Instructions: In the questions below, predict likely structures of metabolites of mepyramine, one of the early antihistamine drugs. Drawing the metabolite structures will require you to watch a video on the use of JSDraw, a chemistry drawing program. Learning Goals: To learn how to use JSDraw and gain experience predicting metabolites. JSDrawtutorialvideo Please watch the online video (4 minutes, 33 seconds). Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. 8.3PhaseIPtII Reductionandhydrolysisvideo Please watch the online video (3 minutes, 55 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. PredictingphaseImetabolites Background: One can often predict the metabolites of a drug based on the drug's functional groups. Instructions: In the questions below, predict likely phase I structures of metabolites of the indicated drugs. Learning Goal: To gain experience predicting phase I metabolites of a drug. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. 8.4PhaseII Conjugationvideo Please watch the online video (6 minutes, 41 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Roleofglutathione Background: Glutathione is a nucleophilic molecule that reactions with electrophilic molecules in the liver. Instructions: Read the passage below concerning how glutathione protects the liver and body from being damaged by certain drugs. Learning Goals: To understand better how the liver metabolizes drugs and the limits of the liver in detoxification. The liver breaks down drugs, primarily through its arsenal of oxidative CYP‐450 enzymes. The process of oxidation involves the loss of electrons. As a molecule loses electrons, it becomes electron poor and therefore more electrophilic. Strong electrophiles, which are often formed through metabolic reactions, can be very damaging to cells. This raises a question... How can the liver continuously perform oxidative reactions on drugs and other molecules without being extensively damaged by electrophilic metabolites? The answer is that the liver is protected by processes such as glutathione conjugation. Glutathione, a natural nucleophile through its thiol group, reacts with strong electrophiles and sacrifices itself before the liver tissue is damaged. The protection of the liver is only limited by the liver's stores of glutathione. The concentration of glutathione in the liver is around 5 mM. If a person ingests too much of a compound that reacts with glutathione, then once the glutathione reserves are consumed, the liver will be damaged. It is for this reason that drug overdoses often lead to extensive liver damage PredictingphaseIImetabolites Background: Based on the functional groups in a molecule, the metabolites of a drug can often be predicted. Instructions: In the questions below, predict likely structures of phase II metabolites of the drugs that are shown. Learning Goal: To gain experience predicting phase II metabolites. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. 8.5MetabolismIssues Metabolisminhibitionvideo Please watch the online video (7 minutes, 24 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Casestudy‐cimetidine Background: Drugs that inhibit any of the CYP enzymes have the potential to interfere with the metabolism of other drugs. This type of drug interaction can cause safety problems and affect the marketability of a drug. Instructions: Read the passage below about CYP inhibition of cimetidine. Learning Goals: To learn about the implications of CYP inhibition of a drug. Cimetidine is a histamine H2‐receptor antagonist and represented a first‐in‐class drug for the treatment of acid reflux. Cimetidine was marketed in the United States in 1979 under the brand name Tagamet. Cimetidine met great success and enjoyed high sales. In 1983 another histamine H2‐receptor antagonist, ranitidine, reached the market in the United States. Ranitidine, sold under the name of Zantac, has a structure that is strikingly similar to cimeditine. Ranitidine is an example of a me‐too drug. Me‐too drugs tend to follow quickly behind a first‐in‐class drug and have similar structures. The composition of matter patents surrounding cimetidine emphasized the importance of the imidazole ring for the activity of histamine H2‐ receptor antagonists. The researchers who discovered ranitidine were able to use a furan ring in place of imidazole and still maintain activity. A me‐too drug rarely matches the profitability of the first drug in a class. The first drug dominates the market, and the me‐too drug lags behind. Ranitidine, however, surpassed the market share of cimetidine in a just a few years. The reason behind the success of ranitidine was CYP inhibition by cimetidine. Cimetidine inhibits several forms of CYP, including CYP1A2, CYP2D6, and, most importantly, CYP3A4. Cimetidine therefore has a long list of drug interactions. While ranitidine also inhibits several CYP forms, ranitidine is a less potent inhibitor than cimetidine. Many patients who experienced complications with cimetidine switched to ranitidine Factorsinmetabolism Background: Based on the functional groups in a molecule, the metabolites of a drug can often be predicted. Instructions: Genetic differences can play a significant role in how a drug is metabolized in one patient or another. Other factors also are important. Learning Goal: To learn how health and age affect drug metabolism. While genetic differences can affect drug metabolism, metabolic differences between people of different races are typically very small, even negligible. Gender is yet another factor that is normally of little importance when discussing metabolism. Other factors, including age and health, are far more influential with regard to drug metabolism. The compound theophylline, a drug sometimes used to treat asthma, highlights these differences.1 Age can greatly influence drug metabolism. Below is a table showing how half‐life can vary with age for theophylline. Bear in mind that theophylline is cleared primarily by the liver, so changes in half‐ life reflect changes in liver activity. age half‐life (h) infants (1‐2 days) 25.7 infants (3‐30 weeks) 11.0 children (1‐4 years) 3.4 children (6‐17 years) 3.7 adults (18‐60 years) 8.7 elderly (>60 years) 9.8 The variability of the half‐life of theophylline shows how metabolism changes with age. Very young infants are not completely developed metabolically, so the half‐life is long. By an age of 12 months, children are metabolically extremely active and the half‐life of theophylline drops below 4 hours. Once a person matures physically, the half‐life lengthens to just under 9 hours. Late in life, as a person's metabolism gradually slows, the half‐life of theophylline also lengthens slightly. Physical health can also affect the half‐life of a drug. Below is a table showing how half‐life can vary with health or physical condition for theophylline. condition half‐life (h) liver cirrhosis 32.0 hepatitis 19.2 pregnancy (1st trimester) 8.5 pregnancy (2nd trimester) 8.8 pregnancy (3rd trimester) 13.0 sepsis 18.8 hypothyroid 11.6 hyperthyroid 4.5 Conditions that negatively affect or place stress on the liver (cirrhosis, hepatitis, late pregnancy, blood infection, and hypothyroid) lengthen the half‐life of theophylline. Hyperthyroidism, a condition in which a patient has an accelerated metabolism (among other things), causes a dramatic decrease in half‐life. Another condition that frequently affects a drug's half‐life is diabetes. Advanced diabetic patients often have limited kidney function. Renal clearance is diminished, and drugs that rely on the kidneys for clearance have lengthened half‐lives. 1. Murray, L., Sr. (Ed.). Physician's Desk Reference (58th ed.) Montvale, NJ: Thomson PDR, 2004. OPTIONAL‐Please participate in the online discussion forum. Pharmacokineticscatter Background: Drugs are listed with distinct pharmacokinetic parameters. These parameters provide a sense of precision for a drug and exactly how it behaves. Instructions: Read the short passage below and review the figures in the linked article (http://aac.asm.org/content/50/7/2281.full) in Antimicrobial Agents and Chemotherapy and answer the questions that follow. Learning Goal: To appreciate the broad variability in the action of drugs across different patients. One of the drugs used to prevent and treat malaria is mefloquine. Mefloquine is administered orally in its racemic form and is well absorbed. The drug is mostly excreted through the bile, meaning it is absorbed, collected in the gall bladder, excreted in bile, and exits the body in feces. Compounds found in the feces are often not absorbed, but in this case, the drug is indeed absorbed from the gastrointestinal tract. In a study on mefloquine dosing, 50 patients received 8 mg mefloquine per kg of body mass (8 mg/kg) on Day 0, Day 1, and Day 2 of the study. Patients were then monitored for approximately one month for their Cp levels of mefloquine. From this data, the pharmacokinetic parameters of mefloquine could be determined. The ideal Cp‐time curve (http://aac.asm.org/content/50/7/2281/F2.expansion.html) shows a smooth, predictable relationship between Cp and time. The scatter plot (http://aac.asm.org/content/50/7/2281/F1.expansion.html) of the experimental data gives a much less convincing relationship based on a visual inspection. The scatter seen in the experimental graph is representative of experimental pharmacokinetic data. The behavior of a drug can vary wildly among different patients. It is for this reason, in part, that drug must have a wide therapeutic window. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. 8.6Prodrugs Prodrugsvideo Please watch the online video (6 minutes, 43 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Casestudy‐antiplateletdrugs Background: Prodrugs are compounds that are converted in the body to the biologically active form of the molecule. Many prodrugs are administered in an inactive form because the active form is not well absorbed. Instructions: Read the passage below about two antiplatelet drugs, clopidogrel and prosugrel. Learning Goal: To understand that metabolic activation of a prodrug can have problematic genetic variability across a broad patient population. Antiplatelet drugs are compounds that inhibit clot formation and therefore reduce the incidence of stroke, heart attacks, and other clot‐dependent conditions. Antiplatelet drugs can fall into several different classes, one of which is the adenosine diphosphate receptor inhibitors. Two examples of adenosine diphosphate receptor inhibitors are clopidogrel and prasugrel. Both clopidogrel and prasugrel are prodrugs. Clopidogrel was approved by the US FDA in 1997. The compound is activated in the body by phase I oxidation of the thiophene ring. One of the sp2 hybridized carbons in the ring is oxidized by CYP2C19. The resulting hydroxythiophene is unstable and hydrolyzes to give the ring‐opened, active form of the drug. Around 5‐10% of the US population have a genetic variation in CYP2C19 that renders the enzyme less active in the metabolism of clopidogrel. For these patients clopidogrel is not activated properly in the body, and the patients do not have therapeutically effective levels of the metabolite. Because of the potential lack of effect of prosugrel, the FDA added labeling in 2010 to clopidogrel packing to indicate the possible lack of effectiveness of the drug. This so‐called black box warning raises safety awareness and concerns for both prescribing physicians and patients. Prasugrel is in the same drug class as clopidogrel and was approved in 2009. Prasugrel is nearly identical in structure to clopidogrel. The major difference is that the thiophene ring already bears an oxygen. The ring is essentially oxidized in its administered form. In the body the drug is activated by hydrolysis of the acetate ester to form the hydroxythiophene metabolite, which forms the active form of the drug. Prasugrel, which is activated by plasma lipases instead of CYP2C19, shows fewer variations in how it is metabolized across broad populations of patients. For this reason, prasugrel is not packaged with black box warnings. Prasugrel is an example of how understanding metabolic issues of a drug allowed the clever design of a safer version of what is otherwise a nearly identical drug. OPTIONAL‐Please participate in the online discussion forum. Drawingprodrugs Background: Prodrugs are compounds that are converted in the body to the biologically active form of the molecule. Instructions: Do the problems below by predicting the structure of the prodrug based on the structure of its active metabolite. Learning Goal: To recognize how particular functional groups can be formed in the body through metabolic reactions. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. Survey Week4Survey Please complete the very brief online survey, which should take less than one minute. Examination2 SecondExamination The exam is open book and open notes. All questions may be attempted once, so be certain of your answer before submitting it. There are ten questions. Each is its own unit within the Examination 2 subsection. Remember that you are bound by the honor code. No postings to the forum concerning the exam are allowed. Furthermore, you must work on the examination independently Problems Please complete the online problems in Examination 2.