Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Heart failure wikipedia , lookup
Mitral insufficiency wikipedia , lookup
Myocardial infarction wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Heart arrhythmia wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Electrocardiography wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
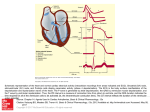
Cardiovascular Research (2013) 97, 182–191 doi:10.1093/cvr/cvs299 Early repolarization in mice causes overestimation of ventricular activation time by the QRS duration Bas J. Boukens1,2*, Mark G. Hoogendijk 2, Arie O. Verkerk 1, Andre Linnenbank 2, Peter van Dam 3, Carol-Ann Remme 2, Jan W. Fiolet 2, Tobias Opthof 2, Vincent M. Christoffels 1, and Ruben Coronel 2 1 Department of Anatomy, Embryology, and Physiology, Academic Medical Center Amsterdam, Amsterdam, the Netherlands; 2Heart Failure Research Center, Department of Experimental Cardiology, Academic Medical Center, Rm. L2-104, Meibergdreef 9, 1105 AZ Amsterdam, the Netherlands; and 3The Donders Institute for Brain, Cognition and Behaviour, Centre for Neuroscience, Radboud University Nijmegen, Nijmegen, the Netherlands Received 23 April 2012; revised 10 September 2012; accepted 14 September 2012; online publish-ahead-of-print 20 September 2012 Time for primary review: 22 days Aims Transgenic mice are frequently used to investigate the role of genes involved in cardiac conduction. The QRS duration calculated from the electrocardiogram (ECG) is a commonly used measure for ventricular conduction time. However, the relation between ventricular activation and QRS duration calculated from a mouse surface ECG is not well understood. We aim to relate ventricular activation and repolarization patterns with the mouse ECG. ..................................................................................................................................................................................... Methods Ventricular activation and repolarization patterns generated by high-density optical mapping and a six-lead pseudoECG were compared in isolated mouse hearts. In addition, mouse ECGs were simulated in silico. Right-ventricular and results activation ends later than left-ventricular activation. Final activation coincided with the end of the QRS complex in leads III and aVF, but not in leads I, II, aVR, and aVL. The pattern of early repolarization (at 20% of repolarization, RT20) but not of RT50 or RT80 followed the activation pattern. After sodium channel blockade by ajmaline, total ventricular activation time increased by 10.0 ms, whereas QRS duration increased by only 2.1 ms. In mice carrying a mutation in Scn5a (1798insD), ventricular activation ended after the end of the QRS complex (12.9 + 0.1 vs. 10.8 + 0.3). ..................................................................................................................................................................................... Conclusion In the mouse, ventricular myocardium activation and early repolarization waves are simultaneously present. This hampers unequivocal interpretation of the duration of the QRS complex as a measure of ventricular activation duration, especially when conduction is slowed. Under these conditions mapping of local activation and repolarization patterns is required for correct interpretation of the ECG. ----------------------------------------------------------------------------------------------------------------------------------------------------------Keywords ECG † Transgenic † Mice † QRS † Repolarization 1. Introduction The murine electrocardiogram (ECG) is often used to study altered cardiac conduction and repolarization resulting from genetic mutations in relation with cardiac arrhythmias and sudden death.1 – 3 Despite substantial differences in the morphology of the ECG in mice and man, similar criteria are used for analysis.2,4 This approach is poorly validated and can lead to misinterpretation of the mouse ECG and, consequently, to incorrect genotype–phenotype relations. On the human ECG, ventricular activation and repolarization can be distinguished as discrete processes because a relatively long period with small changes of the transmembrane potential (the plateau phase of the human action potential) separates the two.5,6 The mouse ventricular action potential lacks a clear plateau phase.4 Thus, repolarization starts when another part of the heart is not yet activated.2,7 Nevertheless, definitions of electrocardiographic deflections and intervals in terms of cardiac conduction8,9 and cardiac repolarization10,11 that are derived from the human ECG are used for the interpretation of the mouse ECG. Application of the criteria for QRS duration in man to the mouse ECG results in marked variation in the QRS duration depending on which lead is selected.12 This variation makes the QRS interval in mice an inaccurate measure for ventricular conduction and renders genotype– phenotype relations cumbersome and questionable. * Corresponding author. Tel: +31 20 566 5367; fax: +31 20 697 6177, Email: [email protected] Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2012. For permissions please email: [email protected]. 183 Understanding the mouse ECG In this study, we determined the relation between the QRS complex and ventricular activation and repolarization during normal and abnormal conductions in mice. Our results show that the end of ventricular activation corresponds only to the end of the QRS complex in leads III and aVF. However, during slow conduction or altered activation patterns, QRS duration, respectively, underestimates or overestimates total ventricular activation time in every lead. method (Figure 1).15 The QT interval was corrected (QTc) according to Mitchell et al.16 2.5 In silico We used a computer model (ECGSIM) based on the structure and activation sequence of the human heart to model the ECG using action potentials of human and mice.17 2.6 Statistics 2. Methods 2.1 Animals The investigation was performed in agreement with national and institutional guidelines and conforms with the European Commission Directive 2010/63/EU and has been approved by the local Animal Experiments Committee, Academic Medical Center. For this study, we used eight adult FVB/N wild-type, three adult 129P2 wild-type, and five adult 129P2 Scn5a1798insd mice.13 2.2 In vivo ECG recording Animals were anaesthetized with 1.5% isoflurane. Electrodes were placed at the right (R) and left (L) armpit and the left groin (F). A reference electrode was placed at the right groin. ECGs were recorded (Biosemi, Amsterdam, the Netherlands; sampling rate 2048 Hz, filtering DC 400 kHz (3 dB)] for a period of 5 min. From these leads, a standard six-lead ECG was calculated as follows: I ¼ L-R, II ¼ F-R, III ¼ F-L, aVR ¼ R-(L+F)/2, aVL ¼ L-(R+F)/2, and aVF ¼ F-(L+R)/2. 2.3 Optical mapping experiments After ECG recording, the mice were killed by cervical dislocation. The heart was rapidly excised, cannulated, mounted on a Langendorff perfusion set-up as described previously.14 During optical mapping, a pseudo-ECG was recorded from electrodes placed in the tissue bath, 5 mm from the heart (Biosemi, Amsterdam, The Netherlands; sampling rate 2000 Hz, filtering DC 400 kHz (3 dB)] in a configuration similar to that of the extremity leads of the standard ECG. The electrodes were placed at the right (R) and left (L) side of the base of the heart and at the left side of the apex (F). Optical action potentials and a pseudo-ECG were simultaneously recorded and the first moment of activation of the right atrium was aligned with the onset of the P-wave. The start of the QRS complex was set to zero. The local moment of activation was defined as the maximum positive dV/dt of the action potential. Three hearts showed delayed left-ventricular activation after placing in the Langendorff set-up. The ventricular activation pattern returned to normal after 10 min. Conduction was slowed by perfusion with 5 mM Ajmaline (Gilurytmalw, Carinopharm). 2.4 ECG criteria The PR interval was defined as the time from the start of the P-wave to the first deflection of the QRS complex. The iso-electric line was defined as the line connecting the end of the T-wave and the start of the P-wave of the next beat. During perfusion of Ajmaline, atrial pacing was necessary and the PR interval was defined as the interval between the start of the stimulus artefact to the start of the QRS complex. The start of the QRS complex was defined as the earliest moment of deviation from baseline in any lead. The end of the QRS complex in each lead was defined as the moment when the S-wave returned to the isoelectric line.8,9 The start of the J-wave was defined as the end of the S-wave. The end of the J-wave was defined as the moment where the positive J-wave turned into the negative T-wave. We determined the end of the T-wave by the tangent The characteristics of the in vivo recorded and the pseudo-ECG were compared using a paired sample t-test. Other group comparisons were performed using one-way ANOVA with Bonferroni test for post-hoc matched pairs. Values are given as mean + SEM. A P-value , 0.05 was considered statistically significant. 3. Results 3.1 Pseudo-ECG is similar to ECG recorded in vivo We recorded an ECG in the intact mouse and compared it with the pseudo-ECG recorded in the Langendorff set-up in each mouse. Figure 1A shows an example of a six-lead ECG recorded in vivo and a six-lead pseudo-ECG of the same heart. Figure 1B illustrates the analysed ECG parameters. The morphology is virtually identical. The table summarizes the electrocardiographic characteristics before and after cardiac isolation. Heart rate was significantly lower after cardiac explantation in accordance with previous findings.18 The other ECG intervals were not statistically different, including the QT interval after correction for the difference in heart rate (Table 1). 3.2 Complete ventricular activation is reflected by the end of the QRS complex in aVF and lead III The duration of the QRS complex varies along the different leads. On average, the differences amounted to about 1.6 + 1.4 ms or 20.9 + 18.9% of the duration of the shortest QRS complex (Figure 2). To determine in which of the leads the end of the QRS complex correctly represents the end of ventricular activation, we reconstructed the ventricular activation pattern and compared the moment of last ventricular activation with the end of the QRS complex calculated from simultaneously recorded pseudo-ECG. Figure 2A and B show an example of the ventricular activation pattern and the simultaneously recorded pseudo-ECG. Epicardial breakthrough occurred at the same time at the right- and the left-ventricular apex. The left ventricle activated within 3.8 ms, whereas the last moment of activation of the right ventricle was at 6.6 ms after the onset of the QRS complex. In Figure 2B, the end of right-ventricular activation is indicated by the dotted line in each of the leads. Note that the end of the QRS complex (indicated by the red star) shows a large variation across the leads, and does not correspond to the end of activation (upstroke of the first and last action potentials shown subsequently). On average, breakthrough occurred simultaneously at the right and left ventricular walls and was 1.6 + 0.4 ms later than the start of the QRS complex (n ¼ 8) (Figure 2C). The final moment of activation was on the right-ventricular myocardium and was significantly later than the last moment of activation of the left-ventricular myocardium (6.7 + 0.4 vs. 4.0 + 0.4 ms, P , 0.05). The end of ventricular activation did not correlate to the end of the QRS complex in any lead 184 B.J. Boukens et al. Figure 1 The pseudo-ECG is similar to the ECG recorded in the in vivo condition. (A) An example of a six-lead ECG recording in vivo (left) and in a Langendorff set-up (right). (B) A magnification of lead I of the ECG recorded in vivo. ecg, ECG. Table 1 ECG vs. pseudo-ECG ECG (n 5 8) Pseudo-ECG (n 5 8) .................................................................................... HR (b.p.m.) 406 + 15 311 + 23* PR (ms) 33.2 + 1.8 34.1 + 1.5 QRS (ms) QT (ms) 8.4 + 0.4 66 + 4.4 7.4 + 0.5 80.9 + 7.6* QTc (ms) 54.4 + 4.3 56.2 + 7.6 QRS was calculated from lead III. The other parameters were calculated from lead I. HR, heart rate. *P , 0.05. (20.3, r , 0.2, ns). However, on average, the QRS duration calculated from lead III and aVF was not statistically different from total activation time, whereas QRS duration calculated from the other leads significantly overestimated total ventricular activation time (Figure 2C). 3.3 Part of the QRS complex contains repolarization The mouse action potential has a larger and faster initial repolarization phase than the human action potential and consequently a plateau at more negative potentials (here defined as ‘low plateau’). To determine how this fast initial repolarization phase interferes with QRS duration, we generated potential maps of subepicardial activation and reconstructed the initial repolarization pattern based on 20% of repolarization (RT20) (Figure 3A). The RT20 pattern followed the activation pattern with a delay of 2–3 ms and ended at around 7.2 ms (n ¼ 8). Furthermore, RT20 started before final activation of the RV base (5.4 + 0.3 vs. 6.7 + 0.4 ms P , 0.05, respectively). The potential map shows that 5 ms after the onset of the QRS, an electrical vector was generated that was directed towards the base (Figure 3B). However, 1 ms later, when repolarization in the apex had already started, two vectors were present with opposite directions, one towards the base and one towards the apex. These vectors will at least in part cancel each other, leading to a slower deflection in the ECG than expected. 3.4 The J-wave solely represents repolarization and not activation We compared local repolarization times with the onset and end of the J-wave. The start of the J-wave varied across leads and was the earliest in aVF and lead III (Figure 3C). As expected, the J-wave started after the last moment of activation. Figure 3C shows that the end of repolarization based on RT20 at the right ventricle, but not the left ventricle, coincided with the onset of the J-wave in lead III 185 Understanding the mouse ECG Figure 2 The last moment of activation on the right ventricle determines the end of the QRS complex. (A) The upper panel shows fluorescent photographs from different view of the mouse heart. The lower panel shows the reconstructed activation patterns. (B) Magnification of the QRS complex recorded simultaneously with optical action potentials. The end of the QRS complex calculated from aVF and lead III corresponds best with the last moment of activation at the right ventricle (blue asterisk). (C) A bar graph representing the relation of the end of ventricular activation with the QRS interval calculated from a simultaneously recorded pseudo-ECG (n ¼ 8). The end (right) ventricular activation corresponds best with the QRS interval calculated from lead III or aVF. The differences with the end of the QRS complex was significant in the other four leads. RV, right ventricle; LV, left ventricle. and aVF. The end of the J-wave coincided with the end of repolarization based on RT50 on the right ventricle but not on the left ventricle. Furthermore, the end of the J-wave did not correlate with RT10, RT20, or RT50 (20.9, r , 0.9, ns) from the left apex or base or the right apex or base. Next we determined the direction of the vector that would result from the local potential maps (Figure 3D) and compared it with the electrical vector of the J-wave (Figure 4A). Both the electrical vector of the J-wave and the vector generated by the potential gradient are directed from the right base to the left-ventricular apex. This suggests that the potential gradient that is present between the right-ventricular base and left-ventricular apex underlies the J-wave. The end of the J-wave is defined as the moment when it passes into the T-wave and corresponded with the moment when the potential was equal throughout the heart (Figure 3D). 3.5 T-wave results from repolarization differences between the left and right ventricles To investigate whether the QT interval in mice gives a good estimate of the ventricular repolarization, we compared repolarization 186 B.J. Boukens et al. left to the right ventricle resulted in a vector (Figure 4B, bottom panel) with the same direction as the vector that generated the T-wave in the ECG (Figure 4A). This suggests that the T-wave in mice results primarily from differences in repolarization between the right and the left ventricles. Figure 4C shows a six-lead pseudo-ECG (from a different heart than that of Figures 2 and 3). Also here, the end of the left-ventricular repolarization coincided with the end of the T-wave. 3.6 QRS duration is not an accurate measure for ventricular conduction time when the conduction pattern is altered To study whether the relation between the end of the QRS complex and the end of ventricular activation time during normal activation sequence remains valid during an altered ventricular activation sequence, we studied hearts with spontaneously delayed left-ventricular activation. Figure 5A shows an example of a heart with normal activation pattern (left) and with delayed left-ventricular activation (right). In hearts with spontaneously delayed activation, left-ventricular breakthrough occurred later than right-ventricular breakthrough (3.2 + 0.4 vs. 1.6 + 0.4 ms after QRS onset, respectively P , 0.05, n ¼ 3). The final ventricular activation time, however, was not later during delayed left-ventricular activation than with a normal activation sequence (6.3 + 0.4 vs. 6.7 + 0.4 ms, ns. n ¼ 3). Remarkably, the simultaneously recorded QRS duration varied from 9.3 + 1.3 to 13.1 + 0.9 ms across leads and was markedly longer than during a normal ventricular activation sequence where the QRS duration varied from 7.5 + 0.6 to 11.4 + 0.5 ms. Thus, during delayed left-ventricular activation, the end of the QRS complexes underestimate the total ventricular activation time in every lead. 3.7 QRS duration is not an accurate measure for ventricular conduction time when conduction is slowed by ajmaline Figure 3 The large initial repolarization starts before activation ends and underlies the J wave. (A) Reconstructed activation (right) and repolarization (left) patterns (RT20) are reconstructed, respectively. (B) The potential maps are reconstructed at 5 and 6 ms after the onset of the QRS complex. (C) A bar graph representing summarized data (n ¼ 8) of RT20 and RT50% on the right and the left ventricle. (D) At 7 ms, only one vector is present that is directed to the apex. At 12 ms, potential is similar throughout the heart. AT, activation time; RT20, repolarization time measured at 20% of repolarization; RV, right ventricle; LV, left ventricle; Au, arbitrary units. moments at the right and left ventricles with the end of the T-wave. Repolarization, based on RT50 and RT80, ended earlier on the right than on the left ventricle (Figures 3C and 4B), but despite this, both correlated significantly with the end of the T-wave (r ¼ 0.66 and r ¼ 0.68, respectively, P , 0.05). The potential gradient from the To investigate whether the QRS duration corresponds to the end of ventricular activation when total activation time prolongs, we slowed conduction by applying a sodium-channel blocker ajmaline to the perfusion solution. Figure 5B shows the reconstructed activation pattern during the perfusion of ajmaline. Sinus rhythm decreased from 345 + 21 to 137 + 36 b.p.m. (P , 0.05). To compare ventricular activation times at similar heart rates, we stimulated on the right atrium (7 Hz). The ‘PR’ interval increased from 36.6 + 8.3 to 65.2 + 11.5 ms (P , 0.05). The QRS duration in lead I increased from 8.5 + 2.2 to 10.9 + 0.5 ms and in aVF from 7.6 + 2.2 to 11.7 + 1.5 ms. The total time of ventricular activation, however, increased from 6.2 + 0.1 to 16.2 + 4.5 ms (Figure 5B dotted line at the right). Thus, the QRS complex ended considerably earlier than the last moment of ventricular activation leading to an underestimation of 22%. 3.8 In silico To provide theoretical support for the idea that the early repolarization vectors interfere with those associated with activation, we calculated the morphology of the body surface ECG by the application of a mouse action potential morphology. We used action potentials of ventricular myocytes isolated from human and mouse hearts that were measured previously in our group19,20 (Figure 5C) and then Understanding the mouse ECG 187 Figure 4 The T-wave is the result of differences in repolarization moments on the left and the right ventricles. (A) A vector cardiogram (upper) based on lead I (X-axis) and aVF (Y-axis) (lower). The colour scale indicates local activation and repolarization moments determined by optical mapping. (B) Reconstructed repolarization pattern (upper right) and a reconstructed potential (lower right) map at 60 ms after the onset of the QRS complex. (C) At the left, a typical example of a QRS-T complex aligned with simultaneously recorded optical action potentials. The end of the T-wave (red asteriks) corresponds with the last moment of repolarization on the left ventricle (blue asteriks). RT80, repolarization time measured at 80% of repolarization; RV, right ventricle; LV, left ventricle; Au, arbitrary units. used these to calculate the body surface ECG under normal conditions and during altered conduction. The introduction of an action potential with a steep initialrepolarization phase resulted in a J-wave on the calculated body surface ECG (Figure 5C) resembling the mouse ECG measured in vivo. When we removed the steep initial-repolarization phase and applied an action-potential shape resembling a human myocyte, the J-wave disappeared on the calculated body surface ECG (blue lines). The latter supports the hypothesis that the fast initial repolarization phase of the mouse action potential underlies the J-wave on the body surface ECG. To exclude the possibility that a low-voltage plateau phase can lead to a J-wave as well, we calculated the body surface ECG based on human action potential with reduced amplitude. This led to a QRS complex with lower amplitude but not to a J-wave (red lines). In lead III, the end of the simulated QRS complex coincided with the last moment of ventricular activation, in line with the experimental data (Figure 2C). The QRS complex in lead aVF ended before the last moment of ventricular activation (Figure 5D). When conduction was slowed by reducing sodium-channel density (Figure 5D), the QRS 188 B.J. Boukens et al. Figure 5 QRS duration misinterprets total ventricular activation time during abnormal conduction. (A) Reconstruction of normal activation pattern (upper left) and the relation between the last moment of activation and the end of the QRS complex (lower left). Reconstruction of the activation pattern during delayed left-ventricular activation (upper middle). (B) Reconstructed activation pattern during slowed conduction (upper right). The QRS complex in lead I and aVF underestimate the total ventricular activation time (lower right). (C) Measured (upper right) and modelled (lower right) action potentials of isolated myocytes from a human and mouse heart measured. (D) The modelled QRS complex based on the mouse action potential during normal and slowed conduction. RV, right ventricle; LV, left ventricle. complex ended before the last moment of ventricular activation in both lead III and aVF, as we have shown experimentally in the presence of ajmaline (Figure 5B). 3.9 QRS duration underestimates total ventricular activation time in transgenic mice carrying the Scn5a 1798insD mutation Mutations in the gene encoding the cardiac sodium channel (SCN5A) cause conduction slowing in patients.21 We measured transgenic mice that carry the 17958insD mutation which is the murine equivalent of the human 1795insD mutation13 to investigate whether the relation between QRS duration and ventricular activation time observed during ajmaline infusion also holds true in a genetic model of conduction slowing (Figure 6). The PR interval was longer in Scn5a(1978insD) mice compared with wild-type (49.6 + 0.8 vs. 40.8 + 8.0, P , 0.05), whereas heart rate was not different. The total QRS duration (lead III) and total ventricular activation time was longer in Scn5a(1978insD) mice compared with wild-type (QRS: 10.8 + 0.3 vs. 7.6 + 0.7; total ventricular activation time: 12.9 + 0.1 vs. 7.8 + 2.0, P , 0.05). However, in Scn5a(1978insD) mice, the QRS complex ended 189 Understanding the mouse ECG before the last moment of ventricular activation (10.8 + 0.3 vs. 12.9 + 0.1, P , 0,05), whereas in wild-type mice it did not (7.6 + 0.7 vs. 7.8 + 2.0, ns). To study the relation between the end of the QRS complex and the end of ventricular activation during more physiological rhythms, we stimulated on the right atrium (120 ms). In the Scn5a(1978insD) mice, this resulted in a second-degree atrioventricular block: Wenckebach type. During the Wenckebach cycle the RR interval decreased and the last moment of ventricular activation occurs progressively later than the end of the QRS complex. Thus, when conduction is slowed due to the sodium-channel mutation, the QRS duration underestimates the last moment of ventricular activation. 4. Discussion Our results demonstrate that in the mouse ventricular myocardium, activation and early repolarization waves are simultaneously present. The end of the ventricular activation correlates best with the end of the QRS in leads III and aVF during normal activation patterns. However, in case of altered ventricular conduction, the duration of the QRS complex in these leads does not accurately reflect total ventricular activation time. Furthermore, our results indicate that the J-wave is the result of a repolarization gradient from the rightventricular base to the right-ventricular apex and the whole left ventricle. Finally, the T-wave in mice is primarily associated with differences between the right- and left-ventricular repolarizations. 4.1 QRS complex Our results on the subepicardial activation pattern of the mouse heart are in line with the previous findings,22 but show an important relation between activation and repolarization sequences and the consequences for the ECG. Although on average the QRS duration in aVF and lead III was no different from total ventricular activation time, we, in contrast to the work of Liu et al.,23 did not find a correlation between the end of ventricular activation and the end of the QRS complex in any lead. The differences can be attributed to the much higher resolution of our measurements than those used by Liu et al.23 (monophasic action potentials) that allowed us to detect the final moments of activation with more precision. For example, we and Nygren et al.,22 but not Liu et al.,23 show that the base of the right ventricle but not that of the left ventricle is the last activated. The reason why we did not find a correlation between the last moment of right-ventricular activation and the end of the QRS complex is due to the fast initial repolarization in mice. In humans, the end of the QRS complex reflects the last moment of ventricular activation and is easy to determine because the activation and repolarization are separated by an iso-electric ST segment although simultaneous activation and repolarization has been reported.24 In mice, however, significant repolarization already starts in large parts of Figure 6 The QRS duration underestimated total ventricular activation time in transgenic mice carrying a in mutation in Scn5a (1798insD). The upper panel shows the last three captured beats before a blocked atrial activation. The lower panel shows that the last ventricular activation (blue star) is later than the end of the QRS complex (red star). Rv, right ventricle; lv, left ventricle; s, stimulus artefact. 190 the heart before the final moment of activation at the right-ventricular base (London2 and our study). 4.2 J-wave Spach et al. showed that a positive ST segment (‘J point’) can be found in a healthy human which is most likely the result of posterior-to-anterior difference in activation or repolarization.24,25 However, in these studies, only body surface measurements were performed which makes it difficult to compare their results with our data. In mice, however, the onset of the J-wave has been related to the end of the QRS complex and the start of repolarization.23 Our data show that repolarization already starts during the QRS complex, before the onset of the J-wave (Figure 3B). Furthermore, it has been reported that the J-wave is composed, at least in part, of ventricular activation.12 We show that ventricular activation ends before the onset of the J-wave in each lead and, therefore, does not interfere with the J-wave. However, when conduction is slowed, the J-wave already starts when parts of the heart are not yet activated and then is partly contaminated with activation. This may lead to an altered morphology of the QRS complex in one lead but not in another. Therefore, it is important to study the morphology of the QRS complex in more than one lead. If the morphology is abnormal in one of the leads then several explanations are possible and reconstruction of the activation and repolarization sequence is required for correct interpretation of the mouse ECG. B.J. Boukens et al. such a short distance in well-coupled myocardium.26,27 Furthermore, the contribution of transmural repolarization patterns to the T-wave on the body surface ECG are debated in general.28,29 The morphology of the calculated QRS complexes differed from the measured QRS complexes. This is likely caused by the use of a computer model based on the structure and activation sequence of the human heart.17 Not only differences in size, but also small structural differences are present between the human and mouse hearts.30 Furthermore, the activation pattern of the septum in mouse differs from that of human.31,32 Thus, the structural and functional differences between human and mouse hearts may have caused the differences in morphology between the modelled and measured QRS complexes. 5. Conclusion Our results show that the last ventricular activation is at the base of the right ventricle and corresponds best, although imperfectly, with the end of the QRS complex in leads III and aVF but not in the other standard limb leads. During slowed conduction, the duration of the QRS complex may underestimate total ventricular activation time. Our data show that in mice with altered cardiac conduction by either pharmacological or genetic causes, mapping of local activation and repolarization patterns is required for correct interpretation of the electrocardiographic changes. 4.3 T-wave Whether the T-wave is an accurate measurement for complete ventricular repolarization in mice is debated.12,23 We show that the repolarization sequence generates an electrical vector that is directed from the left to the right ventricle and underlies the T-wave in mice. Liu et al.23 suggested that in mice, the T-wave is the result of subendocardial– subepicardial and apical –base differences in repolarization times. In both studies, the QT interval was measured during a faster rhythm (400 –600 b.p.m.) than in our study (310 b.p.m.). In our study, the differences between repolarization times on the left and right ventricles became smaller when the heart rate was increased (data not shown). However, even at 310 b.p.m., we could always relate the T-wave to an electrical vector caused by right-to-left differences in repolarization moments. We found that the end of the T-wave correlated with RT80 on the right and left ventricles. Danik et al.12 found no correlation between APD90 and the end of the T-wave. A difference between this study and ours is that we determined the end of the T-wave by the tangent method.15 If we used the moment when the T-wave intersected the isoelectric line as the end of the T-wave, like Danik et al.,12 a correlation with RT80 on the right or left ventricle (data not shown) was absent. It has been shown that the latter method is very inaccurate and subject to interindividual variation. Taken together, repolarization abnormalities in mice should be measured by optical mapping or monophasic action potentials but cannot be determined by the body surface ECG. 4.4 Limitations We did not measure repolarization moments in the subendocardium or intramurally because this is not feasible by optical mapping. Therefore, we cannot exclude the contribution of transmural difference in repolarization moments to the morphology of the T-wave. However, it is not likely that large differences in repolarization times exist over Conflict of interest: none declared. Funding This work was supported by grants from STW (project 10959 to A.L. and P.v.D.) and the Netherlands Heart Foundation (2010B205 and 2008B062 to V.M.C. and R.C. and 2007B018 to R.C.) References 1. Berul CI. Electrophysiological phenotyping in genetically engineered mice. Physiol Genomics 2003;13:207 –216. 2. London B. Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 2001;12:1089 –1091. 3. Wehrens XHT, Kirchhoff S, Doevendans PA. Mouse electrocardiography: an interval of thirty years. Cardiovasc Res 2000;45:231 –237. 4. Goldbarg AN, Hellerstein HK, Bruell JH, Daroczy AF. Electrocardiogram of the normal mouse, Mus musculus: general considerations and genetic aspects. Cardiovasc Res 1968;2:93 –99. 5. Freud GE. Transmembrane potential in isolated human heart. Cardiovasc Res 1972;6: 75 –78. 6. Hoffman BF, Suckling EE. Cellular potentials of intact mammalian hearts. Am J Physiol 1952;170:357 –362. 7. Richards AG, Simonson E, Visscher MG. Electrocardiogram and phonogram of adult and newborn mice in normal conditions and under the effect of cooling, hypoxia and potassium. Am J Physiol 1953;174:293 –298. 8. Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet 2010;42:117 –122. 9. Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet 2010;42:149 –152. 10. Salama G, Baker L, Wolk R, Barhanin J, London B. Arrhythmia phenotype in mouse models of human long QT. J Interv Card Electrophysiol 2009;24:77 –87. 11. Brouillette J, Grandy SA, Jolicoeur P, Fiset C. Cardiac repolarization is prolonged in CD4C/HIV transgenic mice. J Mol Cell Cardiol 2007;43:159 –167. 12. Danik S, Cabo C, Chiello C, Kang S, Wit AL, Coromilas J. Correlation of repolarization of ventricular monophasic action potential with ECG in the murine heart. Am J Physiol-Heart Circ Physiol 2002;283:H372 – H381. 13. Remme CA, Verkerk AO, Nuyens D, van Ginneken AC, van BS, Belterman CN et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 2006;114:2584 –2594. Understanding the mouse ECG 14. Aanhaanen WT, Boukens BJ, Sizarov A, Wakker V, de Gier-de VC, van Ginneken AC et al. Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J Clin Invest 2011;121:534 –544. 15. Lepeschkin E, Surawicz B. The measurement of the Q-T interval of the electrocardiogram. Circulation 1952;6:378 – 388. 16. Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol-Heart Circ Physiol 1998;43:H747 –H751. 17. van OA, Oostendorp TF. ECGSIM: an interactive tool for studying the genesis of QRST waveforms. Heart 2004;90:165 –168. 18. Opthof T. Function and structure of the mouse sinus node: nothing you can see that isn’t shown. Cardiovasc Res 2001;52:1 –4. 19. den Ruijter HM, Verkerk AO, Berecki G, Bakker D, van Ginneken AC, Coronel R. Dietary fish oil reduces the occurrence of early afterdepolarizations in pig ventricular myocytes. J Mol Cell Cardiol 2006;41:914 –917. 20. Verkerk AO, den Ruijter HM, de JN, Coronel R. Fish oil curtails the human action potential dome in a heterogeneous manner: implication for arrhythmogenesis. Int J Cardiol 2009;132:138–140. 21. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293 –296. 22. Nygren A, Clark RB, Belke DD, Kondo C, Giles WR, Witkowski FX. Voltage-sensitive dye mapping of activation and conduction in adult mouse hearts. Ann Biomed Eng 2000;28:958 –967. 23. Liu G, Iden JB, Kovithavongs K, Gulamhusein R, Duff HJ, Kavanagh KM. In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J Physiol (Lond) 2004;555:267 –279. 191 24. Spach MS, Barr RC, Benson W, Walston A, Warren RB, Edwards SB. Body surface low-level potentials during ventricular repolarization with analysis of the ST segment: variability in normal subjects. Circulation 1979;59:822–836. 25. Spach MS, Barr RC, Warren RB, Benson DW, Walston A, Edwards SB. Isopotential body surface mapping in subjects of all ages: emphasis on low-level potentials with analysis of the method. Circulation 1979;59:805 – 821. 26. Opthof T, Coronel R, Wilms-Schopman FJG, Plotnikov AN, Shlapakova IN, Danilo P et al. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: Tp-e interval does not reflect transmural dispersion. Heart Rhythm 2007;4: 341 –348. 27. Coronel R, Wilms-Schopman FJG, Opthof T, Janse MJ. Dispersion of repolarization and arrhythmogenesis. Heart Rhythm 2009;6:537 – 543. 28. Oster HS, Taccardi B, Lux RL, Ershler PR, Rudy Y. Noninvasive electrocardiographic imaging: reconstruction of epicardial potentials, electrograms, and isochrones and localization of single and multiple electrocardiac events. Circulation 1997;96:1012 –1024. 29. van OA. Genesis of the T wave as based on an equivalent surface source model. J Electrocardiol 2001;34 (suppl.):217 –227. 30. Rowlatt U. Functional morphology of the heart in mammals. Am Zool 1968;8: 221 –229. 31. Durrer D, Van Dam RT, Freud GE, Janse MJ, Meijler FL, Arzbaecher RC. Total excitation of the isolated human heart. Circulation 1970;41:899 –912. 32. Van Rijen HV, van Veen TA, van Kempen MJ, Wilms-Schopman FJ, Potse M, Krueger O et al. Impaired conduction in the bundle branches of mouse hearts lacking the gap junction protein connexin40. Circulation 2001;103:1591 –1598.